


Cytologie
Les urgences cytologiques en hématologie regroupent un ensemble de situations cliniques critiques, parmi lesquelles les microangiopathies thrombotiques (MAT), les leucémies aiguës, et les fortes leucocytoses. Souvent révélatrices de pathologies graves, elles exigent une prise en charge rapide et coordonnée entre cliniciens et biologistes. Cet article explore ces principales urgences cytologiques, en mettant l'accent sur les anomalies observées sur les frottis sanguins et sur leur rôle central dans l'orientation diagnostique et thérapeutique.
Microangiopathies thrombotiques
Schizocytes
Les schizocytes sont des fragments de globules rouges produits par des dommages mécaniques au sein de la circulation sanguine.
Ils peuvent être retrouvés dans différentes situations, notamment au cours de microangiopathies thrombotiques (MAT). La plus fréquente est le Purpura thrombotique thrombocytopénique (PTT), mais on les rencontre également lors du Syndrome hémolytique et urémique (SHU) et de MAT associées à des contextes particuliers : HELLP syndrome chez la femme enceinte, MAT induites par anticancéreux et autres drogues (Mitomycine C, Gemcitabine, Bortezomib, Ciclosporine, Tacrolimus) ; métastases ; allogreffe de cellules souches hématopoïétiques.
La variabilité de la nature et de la forme des fragments nécessite une appréciation morphologique attentive et rigoureuse. L'International Council for Standardization in Haematology (ICSH) a établi des critères précis pour leur identification et leur décompte (1) :
- Les schizocytes doivent être recherchés au grossissement x40 ou x100 et exprimés en pourcentage des GR totaux après avoir compté au moins 1000 hématies.
- Les schizocytes sont toujours plus petits que des hématies intactes et peuvent prendre la forme de cellules en triangle, en casque ou kératocytes, de cellules en croissant. Ils possèdent au moins 2 angles pointus et des bords nets. La plupart du temps ils présentent un cytoplasme de coloration homogène, sans pâleur centrale (celle-ci peut parfois être légère, moins importante que celle d'un globule rouge normal).
- Les schizocytes peuvent se sphériser et des microsphérocytes (petites hématies rondes de coloration très dense et homogène) peuvent apparaître progressivement au cours des MAT. Les microsphérocytes doivent être pris en compte seulement si les anomalies précédemment citées sont présentes.
- Le seuil de significativité est de 1 % chez l'adulte et le nouveau-né à terme et 5 % chez le nouveau-né prématuré, en l'absence d'autres anomalies morphologiques érythrocytaires ou légères.
Ainsi, la présence d'une anémie hémolytique associée à une thrombopénie doit faire rechercher systématiquement la présence de schizocytes. De même, la recherche des schizocytes doit être entreprise en urgence dès qu'une microangiopathie thrombotique est suspectée car celle-ci peut engager le pronostic vital à court terme.
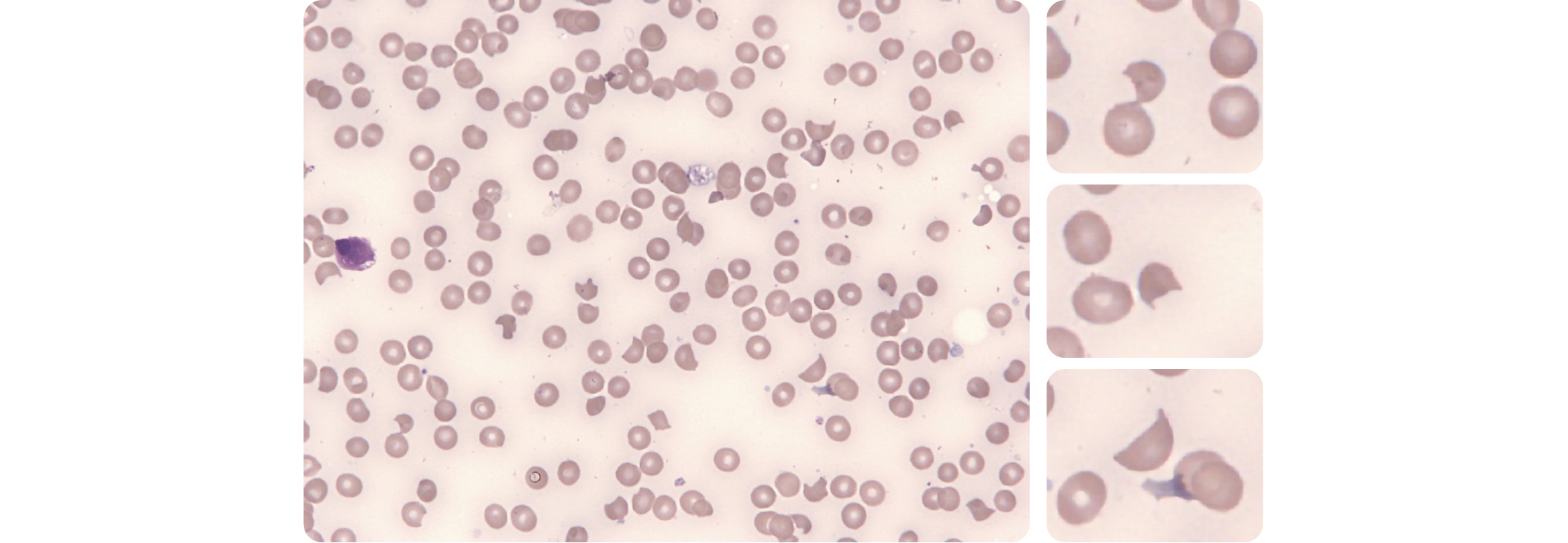
Figure 1 – Observation de schizocytes au frottis sanguin.
La présence d'angles aigus/pointus associée à des bords nets est bien visible.
Leucémies aiguës
Les leucémies aiguës sont un groupe d'hémopathies malignes caractérisées par la prolifération médullaire clonale de précurseurs hématopoïétiques immatures, bloqués dans leur différenciation : les blastes. On compte 4 à 5 cas par an pour 100 000 habitants en France.
On distingue deux types de leucémies aiguës selon l'origine du progéniteur hématopoïétique :
• Les leucémies aiguës myéloïdes (LAM), plus fréquentes chez l'adulte, où prolifèrent des myéloblastes.
• Les leucémies aiguës lymphoïdes (LAL), représentant 80 % des leucémies aiguës de l'enfant, où prolifèrent des lymphoblastes.
Nous traitons ici les principales caractéristiques morphologiques des blastes à rechercher en urgence au frottis sanguin pour orienter le clinicien dans sa démarche diagnostique et thérapeutique.
Classiquement, la NFS associe une anémie, une thrombopénie ainsi qu'une numération leucocytaire allant de la leucopénie à l'hyperleucocytose majeure, avec une blastose sanguine en pourcentage variable.
Leucémies aiguës myéloïdes (LAM)
Les myéloblastes
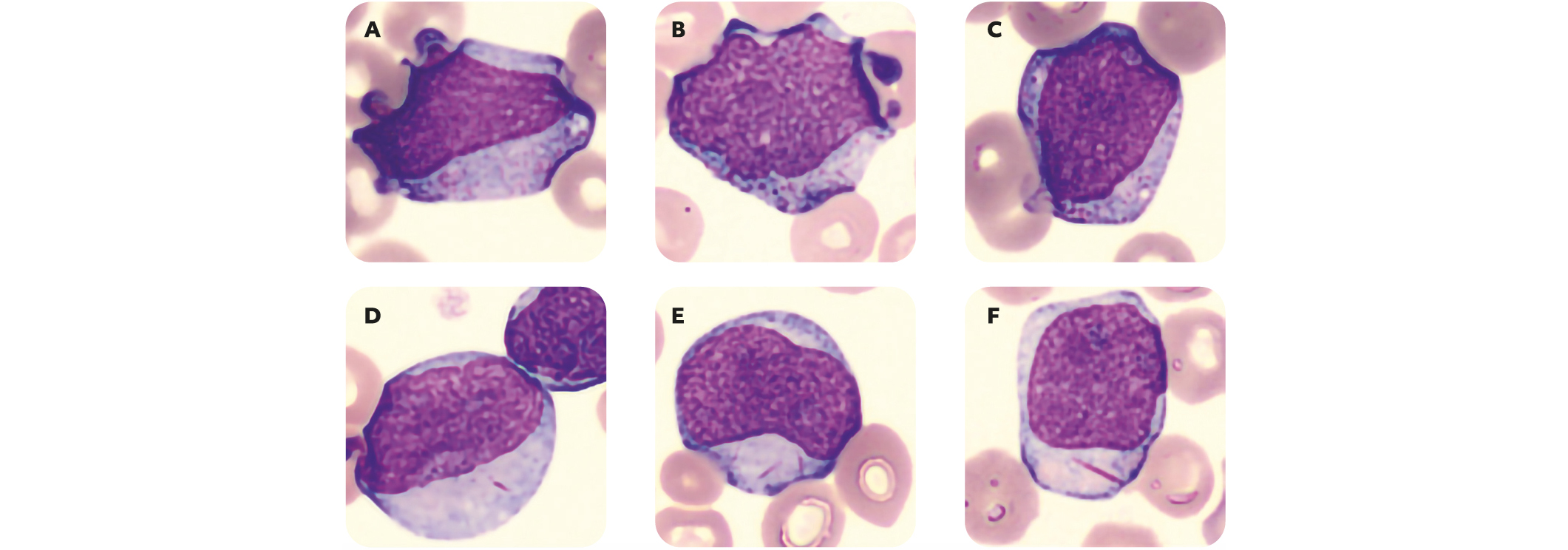
Figure 2 – Myéloblastes présentant des grains en proportion variable
(A-C) et/ou un ou plusieurs corps d'Auer (D-F).
Ce sont des cellules de taille variable, au rapport nucléo cytoplasmique élevé, au contour nucléaire plus ou moins régulier, possédant une chromatine fine et nucléolée, un cytoplasme basophile contenant ou non des granulations azurophiles et un corps d'Auer.
Attention : c'est la présence de corps d'Auer qui permet d'affirmer au clinicien le caractère myéloïde de la blastose. En effet, la distinction entre lymphoblastes et myéloblastes peut parfois être complexe et il existe des LAL dont les blastes peuvent présenter des grains. Dans le cas des LAL, les granulations sont souvent plus grosses et moins nombreuses que dans les LAM.
Qu'est-ce qu'un corps d'Auer ?
C'est une petite structure fine en bâtonnet ou en aiguille issue de la fusion de granulations azurophiles contenant de la myéloperoxydase. Toujours pathologique, le corps d'Auer est spécifique des cellules leucémiques et signe le caractère myéloïde. Il doit donc être recherché avec attention lors de toute suspicion de leucémie aiguë. Le plus souvent unique, on peut en retrouver jusqu'à 3 ou 4 (exceptionnel). La présence de très nombreux corps d'Auer d'aspect « en Fagot » évoque d'emblée une leucémie aiguë promyélocytaire.
La leucémie aiguë promyélocytaire (LAM3)
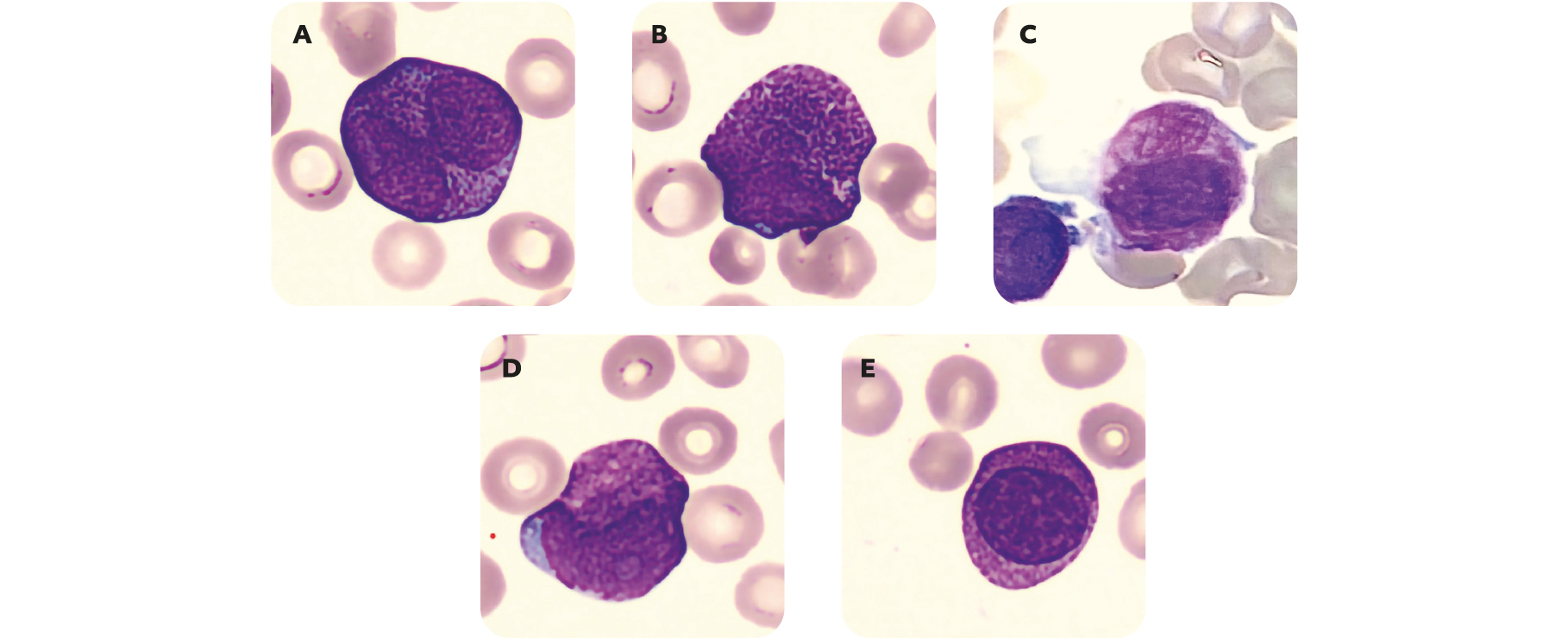
Figure 3 – Aspects cytologiques retrouvés dans la LAM3
(A) Blastes bilobés en bissacs. (B) Blastes hypergranulaires. (C) Blastes avec corps d'Auer en fagot.
(D) Blaste au cytoplasme rosé. (E) Blastes à maturation paradoxale.
La leucémie aiguë promyélocytaire (LAM3 selon FAB) représente environ 5 à 10 % des leucémies aiguës myéloïdes chez l'adulte. Elle survient le plus souvent chez les adultes d'âge moyen (de 30 à 50 ans) et est possible chez l'enfant.
Elle se manifeste souvent par une pancytopénie ou une leucocytose modérée, avec des signes hémorragiques liés à une coagulopathie de consommation. Cependant, il s'agit de la LAM qui a le meilleur pronostic quel que soit l'âge du patient, si une prise en charge rapide et appropriée est initiée (2). Le diagnostic est donc une urgence vitale, car le traitement par ATRA (acide tout-trans-rétinoïque) doit être initié dès la suspicion, avant même confirmation cytogénétique ou moléculaire pour réduire le risque de complications hémorragiques graves.
Le diagnostic cytologique est en première ligne et retrouve différent aspects cytologiques des blastes à différenciation promyélocytaire ou promyélocytes anormaux :
• Blastes bilobés en bissacs.
• Blastes hypergranulaires.
• Blastes avec corps d'Auer.
• Blastes au cytoplasme rosé.
• Blastes à maturation paradoxale.
NB : Les corps d'Auer en fagots ne sont pas nécessaires au diagnostic de LAM3 sur frottis sanguin ou médullaire.
La LAM3 est associée à la translocation t(15;17), menant à la formation du gène de fusion PML-RARα.
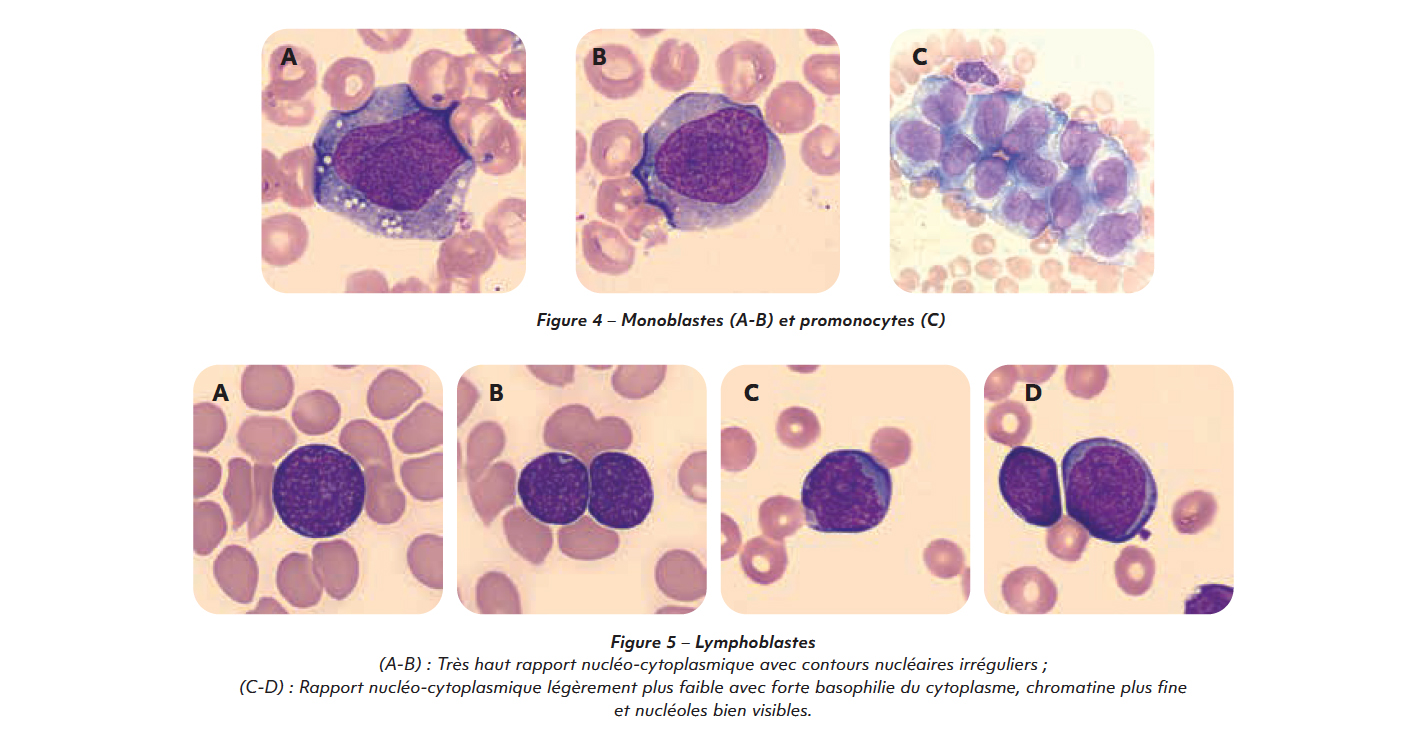
Les monoblastes
Les monoblastes sont des cellules de taille souvent élevée au cytoplasme relativement abondant. Elles possèdent un noyau arrondi ou ovalaire parfois réniforme, une chromatine fine avec souvent un ou plusieurs nucléoles volumineux ainsi qu'un cytoplasme faiblement basophile parfois granulaire et microvacuolé.
La présence de monoblastes peut être associée à celle de promonocytes : ce sont des cellules monocytaires immatures dont le noyau est plus irrégulier que les monoblastes et caractérisé par des replis chromatiniens donnant un aspect cérébriforme. Leur cytoplasme est gris-bleu. Ils peuvent présenter de fines granulations et parfois quelques vacuoles. Les promonocytes sont à inclure parmi les blastes dans la formule sanguine.
Leucémies aiguës lymphoïdes (LAL)
Chez l'enfant, ce sont les blastes dont la taille est la plus petite bien qu'elle reste supérieure à celle d'un lymphocyte. Le plus souvent, les lymphoblastes de l'enfant ont un haut rapport nucléo cytoplasmique (supérieure à 0,9) et sont d'aspect homogène.
Leur noyau présente des contours réguliers, une chromatine fine, un nucléole généralement peu/pas visible, ainsi qu'un cytoplasme modérément basophile dépourvu de granulations et sans vacuoles. La taille et la morphologie des blastes rend leur recherche difficile lorsque le patient est cytopénique.
Chez l'adulte, les lymphoblastes sont généralement de taille moyenne à grande, d'aspect plus hétérogène, au rapport nucléo-cytoplasmique variable (environ 0,8). Leur noyau présente des contours irréguliers, souvent des encoches, un nucléole ou plus, une chromatine fi ne ainsi qu'un cytoplasme de basophilie variable, dépourvu de granulations et sans vacuoles (3).
Diagnostic différentiel : Les Syndromes mononucléosiques (SMN)
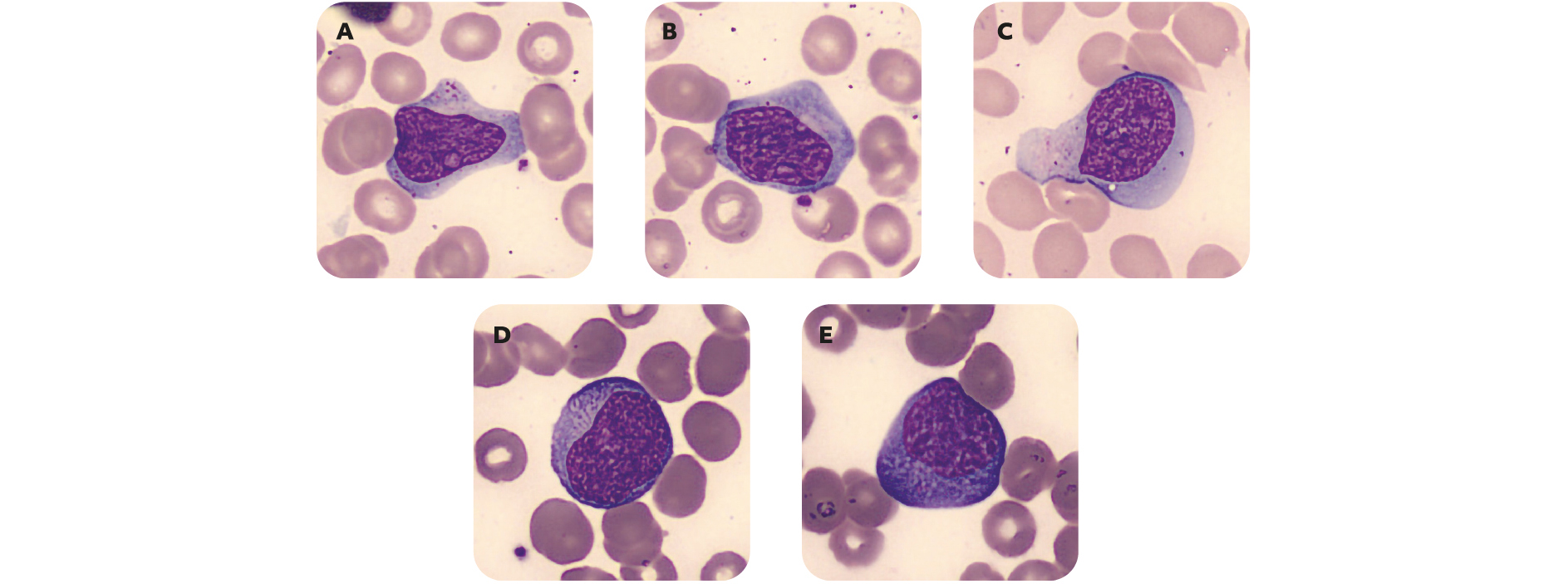
Figure 6 – Lymphocytes activés au cours d'un syndrome mononucléosique
Hétérogénéité morphologique des lymphocytes avec basophilie variable, grains présents ou non, nucléole plus ou moins visible.
Les syndromes mononucléosiques sont un ensemble de pathologies caractérisés par une hyperleucocytose constituée de grands lymphocytes T CD8+ activés. Bien que les signes d'appel du SMN soient évocateurs (asthénie, fièvre, adénopathies, angine et parfois splénomégalie modérée), le tableau clinique peut être bruyant et doit parfois faire écarter une pathologie hématologique maligne.
Cytologiquement, l'hyperleucocytose est souvent modérée (rarement supérieure à 50 G/L) et la présence d'une anémie ou d'une thrombopénie sévère est rare. Par ailleurs, le niveau d'activation variable des lymphocytes se traduit par une grande hétérogénéité morphologique qui permet leur distinction d'autres pathologies.
On retrouve au moins 50 % de lymphocytes au frottis sanguin dont au moins 10 % sont des lymphocytes activés : ce sont des cellules de taille généralement augmentée, au cytoplasme de basophilie légère à intense présentant parfois des grains. Leur noyau montre une chromatine parfois nucléolée qui reste mature.
Hyperleucocytoses hors leucémies aiguës
Lymphome de Burkitt
Le lymphome de Burkitt est un sous type rare de lymphome non hodgkinien agressif à cellules B. Il se manifeste chez les enfants et les adultes et se définit par trois variantes : endémique (africaine), qui se présente principalement sous forme d'une masse maxillaire ou bien abdominale ; sporadique (non africaine) et liée à l'immunodéficience : ces deux formes se manifestent le plus souvent par une masse abdominale.
La forme endémique est liée à l'infection par le virus Epstein-Barr (EBV).
La croissance très rapide de la masse tumorale (indice de prolifération Ki67 supérieur à 95 %) mais aussi la bonne réponse au traitement (taux de guérison 80-90 %) en font une urgence diagnostique et thérapeutique (4).
La forme disséminée du lymphome de Burkitt, auparavant considéré comme une leucémie aiguë lymphoïde, doit faire rechercher des cellules de taille moyenne au rapport nucléo-cytoplasmique élevé (0,8-0,9), au noyau arrondi dont les contours sont réguliers, à chromatine finement mottée présentant 2 à 5 nucléoles. Leur cytoplasme est intensément basophile, dépourvu de granulations, et contient souvent de petites vacuoles.
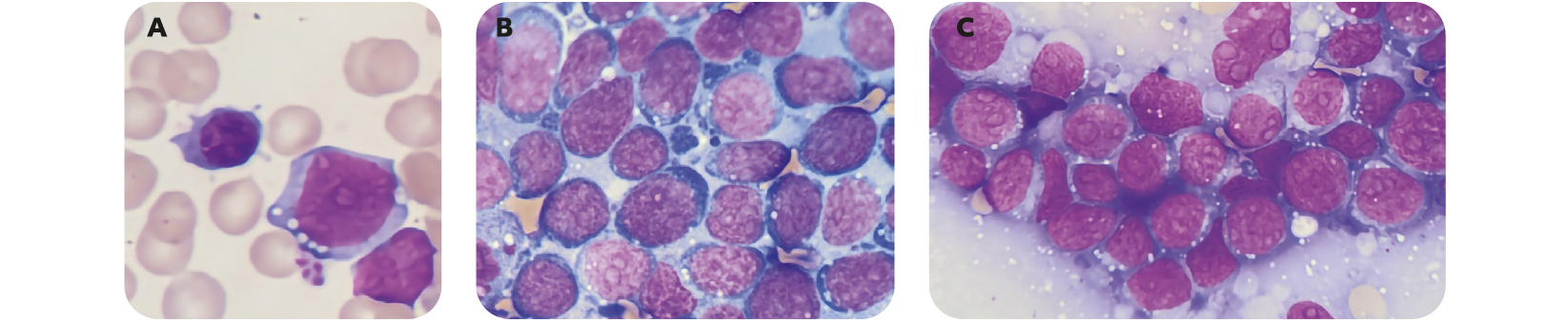
Figure 7 – Cellules lymphomateuses de Burkitt
(A) Frottis sanguin ; (B-C) Apposition ganglionnaire : chromatine finement mottée avec nucléoles bien visibles, nombreuses vacuoles au sein d'un cytoplasme intensément basophile.
Leucémie Lymphoïde Chronique (LLC)
La LLC est la leucémie chronique la plus fréquente de l'adulte occidental, touchant principalement les sujets âgés avec un âge moyen au diagnostic de 70 ans. Le diagnostic est souvent fortuit devant une lymphocytose persistante (supérieur à 5G/L et supérieur à 3 mois). Il s'agit d'une maladie souvent indolente avec une évolution lente, ne nécessitant pas de traitement immédiat dans de nombreux cas.
L'aspect cytologique typique est celui d'une lymphocytose monomorphe composée de petits lymphocytes présentant un rapport N/C élevé, au noyau rond ou légèrement irrégulier, à chromatine dense parfois craquelée et un cytoplasme clair non granuleux.
On retrouve également la présence caractéristique d'ombres de Gümprecht, qui correspondent à des cellules altérées à cause de la fragilité des cellules lors de l'étalement donnant l'impression de noyaux nus.
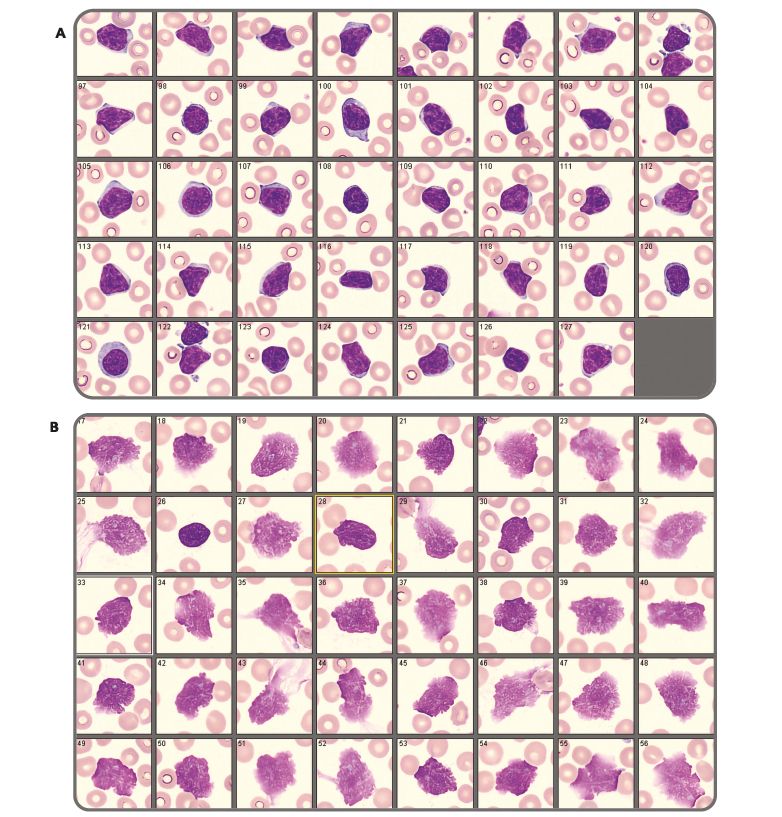
Figure 8 – (A) Hyperlymphocytose (>5G/L) monomorphe essentiellement composée de petits lymphocytes matures à chromatine dense parfois craquelée. (B) Présence de lymphocytes lysés appelés ombres nucléaires ou ombres de Gümprecht sur le frottis.
L'immunophénotypage des lymphocytes sanguins par cytométrie en flux est indispensable au diagnostic. Un score de Matutes de 4 ou 5 est en faveur d'une LLC, caractérisé par l'expression de CD5 et CD23, une expression faible de CD22, l'absence d'expression de FMC7, ainsi qu'une expression faible des immunoglobulines de surface de type kappa ou lambda.
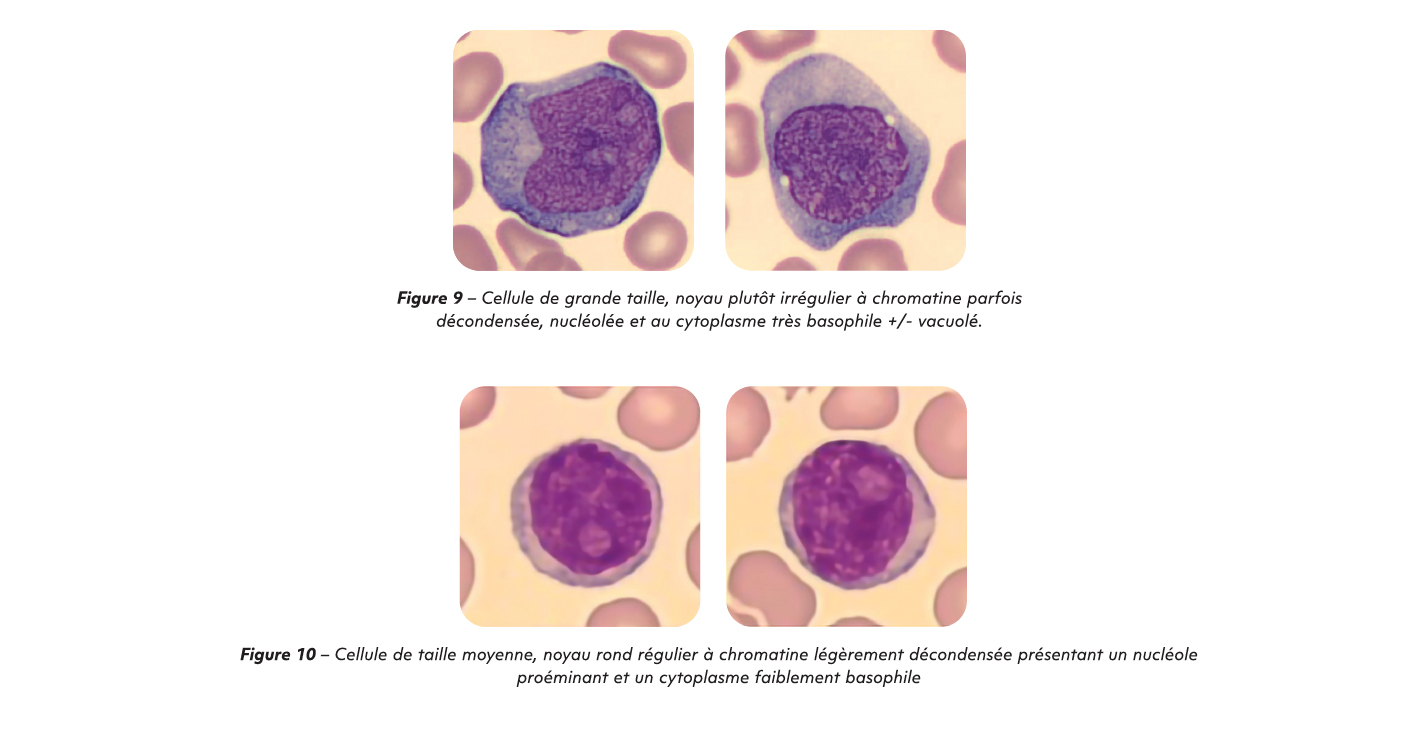
Au cours du suivi de la leucémie lymphoïde chronique (LLC), il est essentiel de rester vigilant face à certaines transformations rares mais graves, pouvant radicalement modifier le pronostic et la stratégie thérapeutique. Parmi celles-ci, le syndrome de Richter correspond à une transformation en lymphome de haut grade (lymphome B diffus à grandes cellules) (5). Il se manifeste cliniquement par l'apparition de symptômes B (fièvre, sueurs nocturnes, perte de poids), un syndrome polyganglionnaire, une hypercalcémie, une augmentation marquée des LDH. Le diagnostic repose sur une biopsie ganglionnaire, révélant un contingent de grandes cellules immunoblastiques. Au niveau de l'hémogramme, la présence des cellules de grande taille, à chromatine décondensée et au cytoplasme très basophile doit être signalée quel que soit le pourcentage car elles peuvent évoquer des cellules de lymphome B diffus à grandes cellules. Cet argument cytologique n'est cependant pas suffisant à lui seul pour conclure au diagnostic étant donné la possible confusion avec des para-immunoblastes qui peuvent parfois être retrouvés en faible quantité dans les LLC classiques. Au niveau de l'immunophénotypage, le score de Matutes est en général faible dans ce contexte, traduisant une perte du phénotype typique de la LLC. Le pronostic est sombre, avec une survie médiane inférieure à 6 mois sans traitement, rendant ce diagnostic une véritable urgence hématologique. La prise en charge thérapeutique repose généralement sur des protocoles de chimiothérapies adaptés aux lymphomes agressifs tel que le R-CHOP. Plus le traitement est initié tôt, meilleures sont les chances de contrôler la progression rapide du lymphome et de stabiliser le patient.
Dans de rares cas, la LLC peut évoluer en leucémie à prolymphocytes (« progression prolymphocytaire de la LLC » selon la classification OMS 2022) qui survient le plus souvent chez le sujet âgé. Elle se caractérise par une hyperleucocytose majeure, dont plus de 55 % sont des prolymphocytes, associée à une anémie et thrombopénie sévères, une AEG, une splénomégalie massive, mais sans adénopathie significative. Là encore, le score de Matutes est bas, témoignant d'un changement dans la biologie de la maladie. Le tableau clinique est généralement brutal et la LPL est peu sensible à la chimiothérapie, avec un pronostic extrêmement péjoratif.
Ainsi, lors du suivi d'une LLC, une attention particulière doit être portée à l'aspect cytologique et à toute modification clinique brutale, afin de ne pas méconnaître une transformation évolutive, dont le diagnostic précoce est crucial pour adapter rapidement la prise en charge thérapeutique.
Leucémie Myéloïde Chronique (LMC)
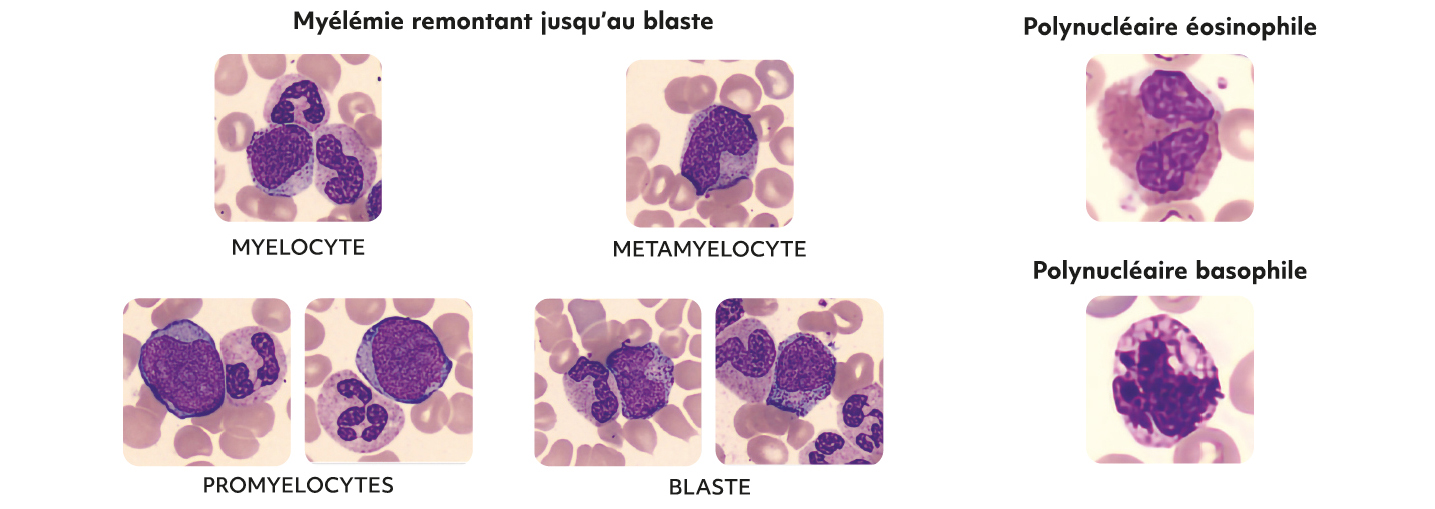
Figure 11 – Aspects cytologiques de la leucémie myéloïde chronique
Myélémie remontant jusqu'au blaste associée à une basophilie constante et le plus souvent à une éosinophilie
La LMC représente environ 15 à 20 % des leucémies de l'adulte et constitue en moyenne 1000 nouveaux cas par an en France avec une prédominance chez l'adulte de plus de 60 ans. Cette hémopathie s'inscrit dans les syndromes myéloprolifératifs chroniques (SMP). Elle est caractérisée par la présence du chromosome Philadelphie issu de la translocation t(9;22). Celle-ci est responsable de la formation du transcrit de fusion (BCR-ABL1) qui code pour une protéine de fusion à forte activité tyrosine kinase, responsable de l'hématopoïèse leucémique. Le diagnostic est souvent posé lors d'un bilan systématique ou en cas de syndrome tumoral (présence fréquente d'une splénomégalie). L'évolution naturelle se fait en trois phases : chronique, accélérée et blastique. Le pronostic s'est considérablement amélioré grâce aux inhibiteurs de tyrosine kinase (TKI) (6).
Au niveau de l'hémogramme, on retrouve souvent une anémie modérée, normochrome, normocytaire et arégénérative souvent accompagnée d'une thrombocytose. Il est également retrouvé une hyperleucocytose franche caractérisée par une polynucléose neutrophile avec une myélémie équilibrée remontant jusqu'au blaste (blastose généralement inférieure à 3 % en phase chronique), une absence de dysgranulopoièse et une basocytose supérieure ou égale à 3 % et +/- une hyperéosinophilie.
Ainsi, devant une hyperleucocytose à PNN non expliquée (infection, inflammation, G-CSF, etc.) avec myélémie chez l'adulte (même jeune), le cytologiste doit rechercher la présence d'une basocytose avec attention, qui orientera le diagnostic vers une LMC.
Références
1. Zini G, d'Onofrio G, Erber WN, Lee SH, Nagai Y, Basak GW, et al. 2021 update of the 2012 ICSH Recommendations for identification, diagnostic value, and quantitation of schistocytes: Impact and revisions. Int J Lab Hematol. 2021;43(6):1264-71.
2. Voso MT, Guarnera L, Lehmann S, Döhner K, Döhner H, Platzbecker U, et al. Acute promyelocytic leukemia: long-term outcomes from the HARMONY project. Blood. 9 janv 2025;145(2):234-43.
3. Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. août 1976;33(4):451-8.
4. Molyneux EM, Rochford R, Griffi n B, Newton R, Jackson G, Menon G, et al. Burkitt's lymphoma. The Lancet. 31 mars 2012;379(9822):1234-44.
5. Parry EM, ten Hacken E, Wu CJ. Richter syndrome: novel insights into the biology of transformation. Blood. 6 juill 2023;142(1):11-22.
6. Senapati J, Jabbour E, Kantarjian H, Short NJ. Pathogenesis and management of accelerated and blast phases of chronic myeloid leukemia. Leukemia. janv 2023;37(1):5-17.

Dr Mathilde IBERTI
Pharmacien biologiste en
hématologie, CHU de Nice

Dr Joy MOUANES-ABELIN
Biologiste médicale en
hématologie
Praticien hospitalier
au CHU de Nice