


Article commenté
Diepstraten ST et al. Putting the STING back into BH3-mimetic drugs for TP53-mutant blood cancers. Cancer Cell. 2024;42(5):850–868.
Introduction
Les altérations de TP53, souvent associées à un caryotype complexe, sont retrouvées dans un large spectre de pathologies hématologiques, incluant les leucémies aiguës myéloïdes (LAM), les syndromes myélodysplasiques (SMD) et certains lymphomes T/NK notamment, et sont caractérisés par l'association à un pronostic défavorable et à une résistance aux thérapies conventionnelles.
En parallèle, les BH3-mimétiques, notamment le venetoclax, ont révolutionné le traitement de certaines hémopathies, en particulier chez les patients atteints de leucémies aiguës myéloïdes (LAM) âgés ou inéligibles à la chimiothérapie intensive (1). En ciblant les protéines anti-apoptotiques BCL-2, ces molécules permettent la libération de protéines pro-apoptotiques telles que BAX, BAK, et induisent alors une perméabilisation mitochondriale externe (MOMP) et la mort cellulaire. Leurs mécanismes d'action sont supposés être p53-indépendants, ce qui laissait espérer une efficacité conservée dans les tumeurs TP53-altérées. Cependant, les observations cliniques et précliniques ont montré que leur efficacité était réduite chez les patients TP53-mutés, suggérant que ces agents nécessitent une activation secondaire de p53 pour produire une réponse apoptotique complète. Face à la résistance liée aux altérations de la voie p53, l'identification de nouvelles vulnérabilités thérapeutiques représente un enjeu majeur (2).
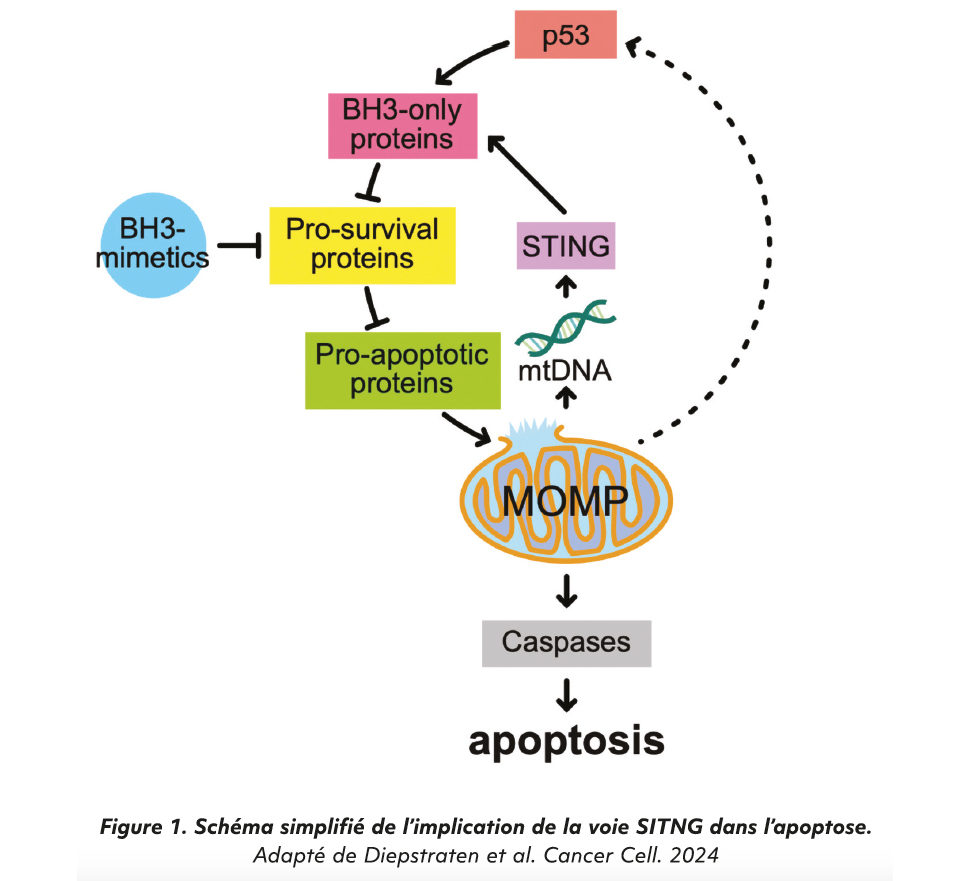
La voie cGAS-STING, initialement décrite comme une sentinelle de l'ADN cytosolique, joue un rôle central dans la réponse immunitaire innée. Son activation déclenche une production d'interférons de type I et de cytokines pro-inflammatoires, via la phosphorylation du facteur de transcription IRF3. Si son rôle en immuno-oncologie a été bien documenté dans les tumeurs solides, en particulier pour améliorer la réponse aux inhibiteurs de checkpoints (3, 4), son implication directe dans l'induction d'une apoptose tumorale autonome reste peu explorée.
C'est dans ce contexte que s'inscrit l'étude de Diepstraten et al. (5), qui propose une nouvelle approche pour surmonter la résistance aux BH3-mimétiques dans les hémopathies TP53-mutées. Les auteurs partent de l'hypothèse que l'activation pharmacologique de STING, en déclenchant une réponse transcriptionnelle via IRF3, pourrait restaurer l'induction des eff ecteurs pro-apoptotiques BH3-only (NOXA, PUMA, BIM) et permettre une re-sensibilisation des cellules TP53-déficientes aux BH3-mimétiques.
Résultats et Discussion
Les auteurs ont dans un premier temps montré que les mécanismes apoptotiques induits par les BH3-mimétiques - venetoclax et S63845 (inhibiteur de MCL-1) - est suivie d'une activation transcriptionnelle de p53 induisant plusieurs eff ecteurs pro-apoptotiques BH3-only, en particulier NOXA, PUMA et BIM ce qui renforce le signal pro-apoptotique initial et fonctionne alors comme une boucle d'intensification de l'apoptose. Ce résultat clarifie un point controversé dans la littérature : bien que le mécanisme d'action des BH3-mimétiques soit en théorie p53-indépendant, leur efficacité in vivo dépend bien de cette amplifi- cation transcriptionnelle secondaire, et plus précisément, c'est l'activation de la MOMP qui serait responsable de l'ampliation via p53.
Cela a été confirmé dans un second temps dans des lignées humaines et des modèles murins de lymphome T/NK inactivés pour TP53 par édition CRISPR-Cas9, avec l'observation de la perte de l'activation transcriptionnelle des gènes BH3-only en présence de BH3 mimétiques, et in fine la réduction de la létalité cellulaire
Dans un deuxième temps, les auteurs ont montré que la MOMP induite par les BH3-mimétiques provoque la libération d'ADN mitochondrial dans le cytosol qui va activer la voie cGAS-STING. Cette activation conduit à la phosphorylation de l'IRF3, un facteur de transcription capable d'induire l'expression de plusieurs gènes impliqués dans la réponse à l'ADN cytosolique, mais aussi dans l'apoptose.
Fait important, les diverses expériences de mort cellulaire montrent que l'eff et de p53 reste le même dans les lignées STING KO.
De plus, dans des cellules TP53- KO, l'activation de STING par des agonistes pharmacologiques (ADU-S100, diABZI, MSA-2) permet une induction partielle mais reproductible de NOXA, PUMA et BIM via IRF3, aboutissant in fine au déclenchement de l'apoptose et à la mort cellulaire, soulignant l'indépendance de la voie STING par rapport à p53 dans sa contribution à l'apoptose.
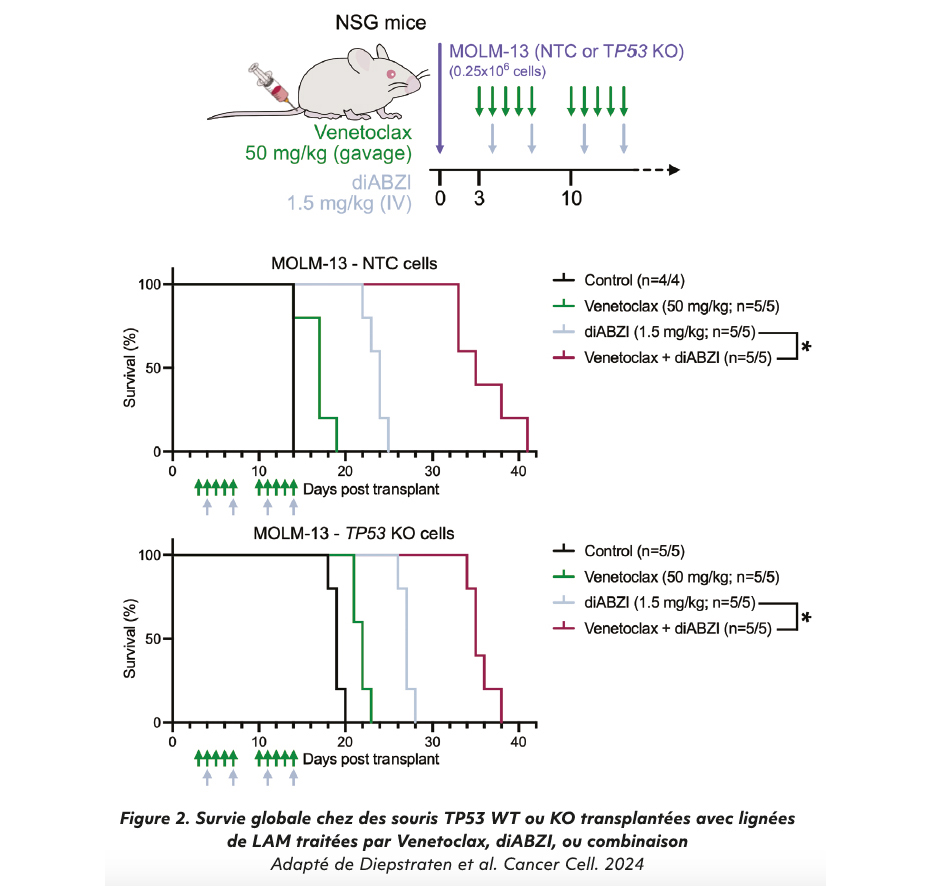
Enfin, les auteurs ont voulu explorer la combinaison des BH3-mimétiques avec les agonistes de STING. Dans les lignées TP53-KO, la combinaison d'un BH3-mimétique et d'un agoniste STING (diABZI) induit une perte de viabilité bien supérieure à celle obtenue avec chaque agent seul. Un point important est que l'eff et des agonistes STING requière un haut niveau d'expression de STING comme c'est le cas dans les lymphomes T/NK extra-nodal ou dans les LAM, mais pas dans les lymphomes B par exemple. Cette stratégie combinatoire a finalement été validée in vivo sur des souris transplantées avec lignées de lymphome T/NL extra-nodal, et avec des lignées de LAM (MOLM-13, Figure 2), ainsi qu'avec des modèles de xénogreff es dérivés de patients.
Conclusion
Cette étude propose un changement de paradigme dans la compréhension de l'action des BH3-mimétiques. Loin d'agir uniquement comme inhibiteurs directs de BCL-2 ou MCL-1, ces molécules induisent, dans les cellules TP53-sauvages, une réponse transcriptionnelle secondaire dépendante de p53, qui amplifie le signal apoptotique via l'induction des eff ecteurs BH3-only. Les auteurs démontrent que cette dépendance à p53 peut être contournée par une activation pharmacologique de la voie STING. En déclenchant la phosphorylation d'IRF3, cette activation induit une expression suffisante de NOXA, PUMA et BIM pour restaurer une apoptose complète. Cette reprogrammation transcriptionnelle pro-apoptotique permet de rétablir la sensibilité des cellules TP53-KO aux BH3-mimétiques, aussi bien in vitro que dans des modèles murins greff és. Le modèle est transposable à des lignées humaines de lymphome NK/T et de LAM ce qui soutient une possible pertinence clinique. Ces résultats ouvrent la voie à des stratégies combinées, fondées non pas sur la génétique mais sur la restauration fonctionnelle des circuits apoptotiques, ce qui pourrait concerner un spectre large d'hémopathies TP53-mutées. La faisabilité clinique de cette approche dépendra d'une part développement de formulations systémiques bien tolérées des agonistes STING, aujourd'hui encore en phase précoce. L'expression hétérogène de STING dans les hémopathies, son impact sur le microenvironnement médullaire et les risques d'inflammation systémique restent également à explorer, même si des confirmations in vivo ont pu être réalisées sur des souris immunocompétentes dans ce travail, sans alerte notable de réaction immunitaire. D'autre part, le seul BH3 mimétique aujourd'hui approuvé reste le vénétoclax. Les inhibiteurs de MCL-1 et de BCL-XL notamment n'ont pour l'instant pas d'autorisation de mise sur le marché faute d'une toxicité notable (6, 7) ce qui rend pour l'instant très hypothétique leur association avec un inhibiteur de STING.
Néanmoins, cette étude s'inscrit dans un courant plus large de repositionnement des agonistes STING comme agents antitumoraux directs. Des essais cliniques dans les tumeurs solides, notamment en combinaison avec des immunothérapies (ex. NCT03172936, NCT03956680), ont déjà montré la faisabilité et les limites de cette approche. Une application dans les hémopathies TP53-mutées pourrait constituer une nouvelle voie de traitement à explorer.
Référence
1. DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. New England Journal of Medicine. 2020;383(7):617-629. doi:10.1056/NEJMoa2012971.
2. Shah MV, Arber DA, Hiwase DK. TP53 ‐Mutated Myeloid Neoplasms: 2024 Update on Diagnosis, Risk‐Stratification, and Management. Am J Hematol. 2025;100(Suppl 4):88-115. doi:10.1002/ajh.27655.
3. Corrales L, Glickman LH, McWhirter SM, et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep. 2015;11(7):1018-1030. doi:10.1016/j.celrep.2015.04.031.
4. Sivick KE, Desbien AL, Glickman LH, et al. Magnitude of Therapeutic STING Activation Determines CD8+ T Cell-Mediated Antitumor Immunity. Cell Rep. 2018;25(11):3074-3085.e5. doi:10.1016/j.celrep.2018.11.047.
5. Diepstraten ST, Yuan Y, La Marca JE, et al. Putting the STING back into BH3-mimetic drugs for TP53-mutant blood cancers. Cancer Cell. 2024;42(5):850-868.e9. doi:10.1016/j.ccell.2024.04.004.
6. Roberts AW, Seymour JF, Brown JR, et al. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J Clin Oncol. 2012;30(5):488-496. doi:10.1200/ JCO.2011.34.7898.
7. Wei AH, Roberts AW, Spencer A, et al. Targeting MCL-1 in hematologic malignancies: Rationale and progress. Blood Reviews. 2020;44:100672.

Jules HIGUÉ
Interne en Hématologie
au CHU de Toulouse
Actuellement en Master 2 au
Walter and Eliza Hall Institute of
Medical Research, Peter McCallum
Cancer Center de Melbourne