


La drépanocytose et plus précisément les syndromes drépanocytaires majeurs forment un groupe de pathologies d'origine génétique héréditaire, de transmission autosomique récessive, affectant le gène codant pour la bêta-globine situé sur le chromosome 11. Il s'agit de la pathologie génétique la plus fréquente au monde avec une incidence qui prédomine sur le continent africain et en Asie du Sud-Est. Son incidence augmente en France et dans le monde16, l'espérance de vie en occident est toujours réduite9 et la mortalité chez les adultes jeunes reste élevée.
Épidémiologie
Le Protocole National de Diagnostic et de Soins (PNDS) attribue une place majeure à la recherche de la drépanocytose dans le programme national de dépistage néonatal au troisième jour, dans l'éventualité où l'enfant fait partie des populations exposées à cette maladie.
En effet, la distribution géographique de la maladie concerne essentiellement les personnes d'origine africaine, antillaise et certaines parties du subcontinent indien. On observe une colocalisation avec le paludisme dont l'expression hétérozygote AS protège des formes graves, en revanche les formes homozygotes drépanocytaires aggravent le pronostic lors d'une infection à Plasmodium spp 4, 10.
En métropole, le dépistage est ciblé mais systématique dans les DROMCOM. En 2022, on retrouvait alors 1 dépistage positif sur 424 nouvelles naissances dans les DROM-COM contre 1 dépistage positif sur 613 en métropole. Ce dépistage permet de mettre en évidence les syndromes drépanocytaires majeurs, les syndromes bêta-thalassémiques ainsi que des hétérozygoties avec une chaîne d'hémoglobine anormale associée à une chaîne normale 2.
En novembre 2022, la HAS recommande la généralisation du dépistage de la drépanocytose à la naissance. Celle-ci sera mise en place au 1er octobre 2024 dans tous les CRDN.
Biologie de la drépanocytose
La forme physiologique majoritaire de l'hémoglobine : l'hémoglobine A (HbA), est formée par un tétramère de protéines avec deux chaînes d'alpha-globine et deux chaînes de bêta-globine (α2β2).
L'hémoglobine S (HbS), anormale, résulte d'une mutation de substitution T→A sur le dix-septième nucléotide du gène de la bêta-globine sur le chromosome 11, entraînant le remplacement d'un acide glutamique (acide aminé chargé) par une valine (acide aminé neutre). Au stade hétérozygote, un sujet atteint de drépanocytose est souvent asymptomatique. En revanche, un patient homozygote est à surveiller car à risque de complications liées à une malformation érythrocytaire14.
En situation d'hypoxie, la protéine d'hémoglobine HbS se polymérise, altérant la forme et les propriétés mécaniques des érythrocytes et entraînant une dégradation de l'hémo-rhéologie. Il en résulte une diminution de l'oxygénation sanguine et de la perfusion tissulaire ainsi que des obstructions capillaires responsables des complications de la maladie. L'acidose, l'hyperthermie ou la déshydratation sont d'importants facteurs de risque de complication.
La substitution de l'acide glutamique par une valine diminue la solubilité de l'HbS et conduit cette dernière à s'associer avec d'autres acides aminés d'une poche hydrophobe sur la chaîne β adjacente. De longs polymères d'HbS s'assemblent ainsi en bandes parallèles pour former une structure hélicoïdale5. Après une période de latence conditionnée principalement par la concentration en HbS, la pression partielle en oxygène, la concentration en 2,3 DPG et le pH au sein de l'hématie, débute la double nucléation et l'élongation du noyau pour former le polymère d'HbS3.
Les fibres d'HbS rigidifient les cellules et tendent la membrane érythrocytaire par modification du cytosquelette entraînant une forme particulière, plus fragile et la survenue de crises vaso-occlusives.
La durée de polymérisation conditionne la déformation de l'hématie et occasionne une importante poïkilocytose (fig1). La falciformation est par la suite entretenue par des perturbations hydroélectrolytiques érythrocytaires1 .
Toutes ces modifications réduisent la durée de vie de l'hématie de cent vingt jours à une quinzaine de jours pour un drépanocyte14.
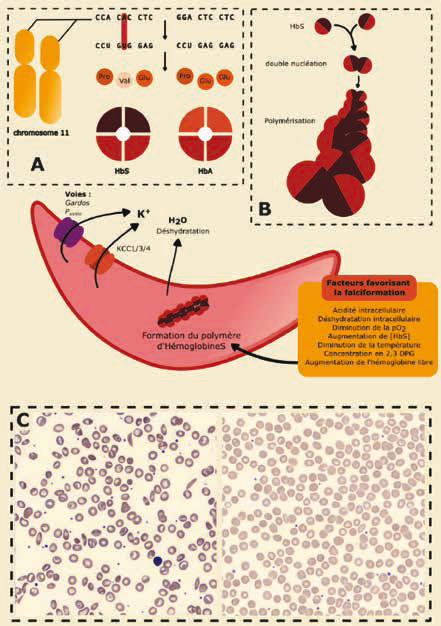
Figure 1 A : Conséquence de la substitution T ->
A sur le gène de la β globine sur le chromosome 11 et formation de l'hémoglobine S (HbS).
B : Formation des polymères d'HbS à partir des monomères grâce à la valine apolaire.
C : Comparaison de deux frottis sanguins entre un patient drépanocytaire à gauche et un patient ne présentant pas de mutation sur le gène de la β globine à droite. Au centre : Falciformation d'une hématie et facteurs influençant la polymérisation de l'hémoglobine S. D'après 7.
Complications de la drépanocytose
La gravité phénotypique clinique dépend du génotype du patient, de son âge et de ses éventuelles comorbidités. En effet, les patients porteurs d'un génotype homozygote SS ou Sβ° ont un risque de complications sévères et récurrentes plus élevé, et un risque de mortalité précoce à l'âge adulte.
Les complications de la drépanocytose, aiguës et chroniques, sont illustrées en Figure 2. Nous présenterons les plus fréquentes et les plus graves6.
Crises vaso-occlusives
La complication la plus fréquente de la drépanocytose est la crise vaso-occlusive qui se manifeste par un accès brutal de douleurs, osseuses ou non, d'intensité très importante. Ces douleurs sont liées à l'obstruction des capillaires dans différents organes qui entraîne une ischémie et éventuellement, après une période prolongée, une nécrose.
La prise en charge est souvent hospitalière en cas d'atteinte de certains organes (cardiomégalie, rétinopathie, insuffisance rénale, etc. (Figure 2)) ou d'échec de la prise en charge ambulatoire. La prise en charge à domicile comporte un repos dans un endroit chaud avec une hydratation quotidienne per os de 2 à 3 litres et la prise d'antalgiques de palier I ou II. En cas d'échec à 24h de cette stratégie, d'isolement social ou de gravité, le recours hospitalier s'impose dans lequel un bilan à la recherche d'un foyer infectieux associé, l'initiation d'antalgiques morphiniques par voie intraveineuse et une prévention de la thrombose par Héparine de Bas Poids Moléculaire (HBPM) seront réalisés. Il est important d'évaluer la gravité clinique de la crise et sa tolérance en recherchant une défaillance hémodynamique ou d'organes.
Syndrome Thoracique Aigu, AVC et infarctus osseux ou splénique
Le syndrome thoracique aigu est la seconde cause de recours hospitalier et la première cause de mortalité dans la drépanocytose car la vaso-occlusion pulmonaire compromet l'oxygénation tissulaire. Il se manifeste une douleur thoracique, une dyspnée, une anomalie auscultatoire, ou une fièvre et peut se manifester par un infiltrat radiologique.
Les accidents vasculaires cérébraux (AVC) sont également fréquents chez les patients drépanocytaires, particulièrement chez les enfants. Les signes cliniques sont très variables (paralysie, céphalées, aphasie, trouble de l'équilibre…) et sont souvent transitoires mais peuvent laisser de graves séquelles intellectuelles et/ou motrices.
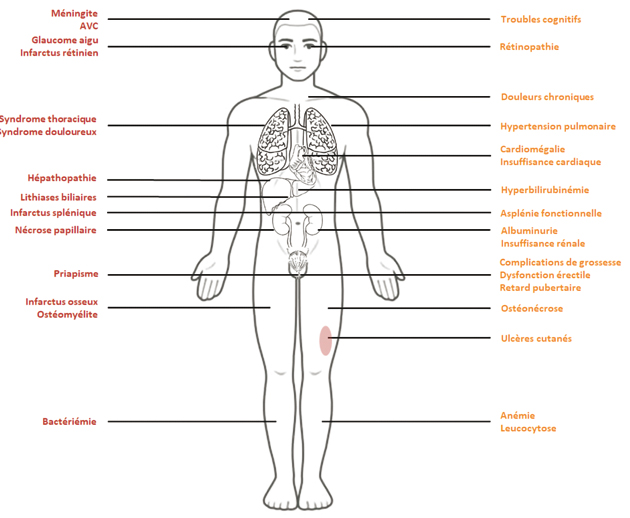
Figure 2 : Complications aiguës et chroniques de la drépanocytose7.
Enfin, la répétition des crises vaso-occlusives peut in fine aboutir à une nécrose tissulaire en particulier osseuse ou organique souvent splénique.
Splénomégalie et anémie
Contrairement aux érythrocytes normaux dont la durée de vie avoisine quatre mois, les érythrocytes drépanocytaires ont une durée de vie d'une quinzaine de jours, ayant pour conséquence une anémie chronique.
Les patients drépanocytaires présentent une hémoglobine se situant entre 70 g/L et 90 g/L, elle peut être plus élevée chez les individus hétérozygotes composites SC et Sβ+. Toute baisse de plus de 20 g/L de l'hémoglobine définie une anémie aiguë dont la cause est le plus souvent une séquestration splénique aiguë liée à une majoration de la destruction des globules rouges anormaux.
L'infection par le parvovirus B19, virus à ADN monocaténaire, entraîne chez le sujet indemne d'hémoglobinopathie une suppression transitoire de l'érythropoïèse qui est, en général, peu profonde et peu symptomatique. En revanche, chez les patients drépanocytaires, l'anémie peut se majorer et nécessiter une hospitalisation pour transfusion. Chez les populations à risque, l'infection par Plasmodium peut également être en cause. Les autres causes d'anémie, carentielle ou inflammatoire, peuvent se surajouter.
Il conviendra d'être attentif à l'hémochromatose secondaire chez les patients polytransfusés.
De nombreuses autres causes d'aggravation de l'anémie pourraient être citées, selon leur origine centrale ou périphérique12.
Infections
Les jeunes enfants atteints de drépanocytose sont les plus exposés au risque infectieux. Celui-ci est lié à une augmentation de la séquestration splénique érythrocytaire et une diminution de la fonction splénique qui altère le fonctionnement du système immunitaire. Les infections les plus fréquentes sont les angiocholites, les cholécystites, les infections urinaires, ostéo-articulaires et pulmonaires.
Il est nécessaire de proposer une prophylaxie quotidienne avec une pénicilline (Oracilline généralement), jusqu'à l'âge de 5 à 6 ans, en absence de contre-indication.
Un respect rigoureux de la stratégie de vaccination est important afin de prévenir en particulier les infections bactériennes.
Ces mesures permettent également de réduire l'occurrence des crises vaso-occlusives qui sont favorisées par un état septique.
Priapisme
Le priapisme est une complication fréquente de la drépanocytose qui touche parfois les enfants et près d'un homme adulte sur deux et se caractérise dans sa forme aiguë par une absence de détumescence pénienne au-delà d'une heure. Il nécessite une prise en charge en urgence pour éviter une ischémie puis une sclérose des corps caverneux et in fine une impuissance irrémédiable et des troubles psychologiques. L'éducation thérapeutique des patients masculins, mêmes jeunes, est essentielle pour éviter les complications. La prise en charge thérapeutique repose sur la prise orale de 20 à 50 mg d'Etiléfrine si la tumescence perdure depuis moins d'une heure, dans le cas contraire de son injection en intra-caverneuse. En cas d'échec après deux injections séparées de 20 minutes il convient de recourir à une prise en charge hospitalière pour un drainage doux et en dernier recours un geste chirurgical.
Ulcères cutanés
Les ulcères cutanés sont une complication parfois invalidante de la drépanocytose. En effet, ils sont le plus souvent localisés dans la région péri-malléolaire ce qui peut limiter la mobilité. Il existe deux formes distinctes : des ulcères de petite taille, cicatrisant en quelques semaines ou mois, et de grands ulcères « malins », pouvant persister plusieurs années.
Traitements de la maladie
Le seul traitement curatif de référence actuellement disponible est l'allogreffe de cellules souches hématopoïétiques avec donneur familial et post conditionnement myéloablatif par Busulfan, Cyclophosphamide et anti-thymoglobuline. Cependant, il s'agit d'un traitement très lourd nécessitant un bon état général et avec des effets secondaires nombreux et parfois graves, en particulier la réaction du greffon contre l'hôte (GVH). Pour cette raison, ce traitement est réservé aux enfants présentant des formes graves de la maladie.
L'hydroxycarbamide, ou hydroxyurée, est un traitement préventif de première ligne des complications vaso-occlusives de la drépanocytose et est indiquée dès l'âge de 2 ans. Son action permet d'accroître la concentration d'hémoglobine fœtale et de réduire les formes à risque de polymérisation composées d'hémoglobine HbS.
Il n'existe pas d'autres traitements préventifs des crises vaso-occlusives. En effet, l'EMA a recommandé de ne pas autoriser le lévoglutamide (L-glutamine), censé réduire les effets du stress oxydatif, en raison d'une absence de démonstration de son efficacité et le crizanlizumab, un anticorps monoclonal bloquant la P-sélectine a vu son AMM retirée en 2023 suite à l'étude de phase III STAND qui n'a pas montré de bénéfice clinique15.
Réservé au traitement de l'anémie hémolytique sévère, le Voxelotor, un inhibiteur de la polymérisation de l'hémoglobine S, est indiqué chez des patients âgés de plus de 12 ans.
De nouvelles pistes prometteuses sont toujours à l'étude avec la thérapie génique qui connaît un essor important depuis la fin des années 201013. En pratique, deux techniques sont utilisées, la plus classique consiste en l'addition d‘un gène dans les cellules cibles via un vecteur viral, la seconde est l'édition du génome par modification d'un gène impliqué dans la maladie avec utilisation par exemple de CRISPR-Cas9. Très récemment, une nouvelle technologie est à l'étude : le “base editing” qui permet des modifications ponctuelles des séquences géniques sans passer par la cassure de la séquence cible11. Les essais cliniques les plus récents et avancés ont validé l'efficacité de ces techniques. Il manque cependant du recul pour évaluer la durabilité et l'impact des modifications géniques de la lignée érythrocytaire et d'éventuels effets secondaires.
Conclusion
Les syndromes drépanocytaires sont des pathologies génétiques fréquentes dont les conséquences cliniques peuvent rapidement être graves et imposer le recours au système hospitalier. L'éducation thérapeutique est fondamentale dans la prise en charge du patient pour permettre une évaluation précoce de la gravité, une initiation rapide des thérapeutiques à domicile et une connaissance des complications liées à cette pathologie. La recherche fondamentale et clinique est très active et de nouvelles thérapeutiques innovantes sont en cours d'évaluation.

Julien CLIQUOT1, 2
Interne de Biologie médicale
CHU de Reims

Arthur COSTE3, 4
Interne en Hématologie
CHU de Reims
Relecture
Emmanuelle GUILLARD1
1 Service d'hématologie biologique et de Biochimie, Pôle de Biologie Territoriale, CHU de Reims.
2 Faculté de Médecine, Université de Reims Champagne Ardennes.
3 Service d'onco-hématologie, Hôpital Robert Debré, CHU de Reims.
4 Faculté de Pharmacie, Université de Reims Champagne Ardennes.
Références
1. Brugnara C, De Franceschi L, Alper SL. Inhibition of Ca(2+)-dependent K+ transport and cell dehydration in sickle erythrocytes by clotrimazole and other imidazole derivatives. J Clin Invest [Internet]. 1993 Jul 1 [cited 2024 Jul 24];92(1):520–6. Available from: http://www.jci.org/articles/view/116597
2. Centre National de Coordination du Dépistage Néonatal. Programme National du Dépistage Néonatal [Internet]. 2021. Available from: https://depistage-neonatal.fr/wp-content/uploads/2023/01/Rapport-Activite-2021.pdf
3. Eaton WA. Hemoglobin S polymerization and sickle cell disease: A retrospective on the occasion of the 70th anniversary of Pauling's Science paper. American J Hematol [Internet]. 2020 [cited 2024 Jul 24];95(2):205–11. Available from: https://onlinelibrary.wiley.com/doi/10.1002/ajh.25687
4. Eleonore NLE, Cumber SN, Charlotte EE, Lucas EE, Edgar MML, Nkfusai CN, et al. Malaria in patients with sickle cell anaemia: burden, risk factors and outcome at the Laquintinie hospital, Cameroon. BMC Infect Dis [Internet]. 2020 [cited 2024 Jul 24];20(1):40. Available from: https://bmcinfectdis.biomedcentral.com/articles/10.1186/s12879-019-4757-x
5. Galkin O, Pan W, Filobelo L, Hirsch RE, Nagel RL, Vekilov PG. Two-Step Mechanism of Homogeneous Nucleation of Sickle Cell Hemoglobin Polymers. Biophysical Journal [Internet]. 2007 [cited 2024 Jul 24];93(3):902–13. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0006349507713474
6. Habibi A, Arlet JB, Stankovic K, Gellen-Dautremer J, Ribeil JA, Bartolucci P, et al. Recommandations françaises de prise en charge de la drépanocytose de l'adulte : actualisation 2015. La Revue de Médecine Interne [Internet]. 2015 [cited 2024 May 31];36(5):5S3–84. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0248866315600029
7. Kato GJ, Piel FB, Reid CD, Gaston MH, Ohene-Frempong K, Krishnamurti L, et al. Sickle cell disease. Nat Rev Dis Primers [Internet]. 2018 Mar 15 [cited 2024 Jun 16];4(1):18010. Available from: https://www.nature.com/articles/nrdp201810
8. Kavanagh PL, Fasipe TA, Wun T. Sickle Cell Disease: A Review. JAMA [Internet]. 2022 Jul 5 [cited 2024 May 31];328(1):57. Available from: https://jamanetwork.com/journals/jama/fullarticle/2793821
9. Lubeck D, Agodoa I, Bhakta N, Danese M, Pappu K, Howard R, et al. Estimated Life Expectancy and Income of Patients With Sickle Cell Disease Compared With Those Without Sickle Cell Disease. JAMA Netw Open [Internet]. 2019 Nov 15 [cited 2024 Jun 1];2(11):e1915374. Available from: https://jamanetwork.com/journals/jamanetworkopen/fullarticle/2755485
10. Luzzatto L. SICKLE CELL ANAEMIA AND MALARIA. Mediterr J Hematol Infect Dis [Internet]. 2012 Oct 3 [cited 2024 Jul 24];4(1):e2012065. Available from: http://www.mjhid.org/index.php/mjhid/article/view/2012.065
11. Mayuranathan T, Newby GA, Feng R, Yao Y, Mayberry KD, Lazzarotto CR, et al. Potent and uniform fetal hemoglobin induction via base editing. Nat Genet [Internet]. 2023 [cited 2024 Jun 3];55(7):1210–20. Available from: https://www.nature.com/articles/s41588-023-01434-7
12. Naik RP, Smith-Whitley K, Hassell KL, Umeh NI, De Montalembert M, Sahota P, et al. Clinical Outcomes Associated With Sickle Cell Trait: A Systematic Review. Ann Intern Med [Internet]. 2018 Nov 6 [cited 2024 Jul 24];169(9):619. Available from: http://annals.org/article.aspx?doi=10.7326/M18-1161
13. Ribeil JA, Hacein-Bey-Abina S, Payen E, Magnani A, Semeraro M, Magrin E, et al. Gene Therapy in a Patient with Sickle Cell Disease. N Engl J Med [Internet]. 2017 Mar 2 [cited 2024 Jun 3];376(9):848–55. Available from: http://www.nejm.org/doi/10.1056/ NEJMoa1609677
14. Steinberg MH. 6 Pathophysiology of sickle cell disease. Baillière's Clinical Haematology [Internet]. 1998 [cited 2024 Jul 24];11(1):163–84. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0950353698800747
15. Study of Two Doses of Crizanlizumab Versus Placebo in Adolescent and Adult Sickle Cell Disease Patients (STAND). Identifier NCT03814746. https://clinicaltrials.gov/study/NCT03814746
16. Thomson AM, McHugh TA, Oron AP, Teply C, Lonberg N, Vilchis Tella V, et al. Global, regional, and national prevalence and mortality burden of sickle cell disease, 2000–2021: a systematic analysis from the Global Burden of Disease Study 2021. The Lancet Haematology [Internet]. 2023 [cited 2024 Jul 5];10(8):e585–99. Available from: https://linkinghub.elsevier.com/retrieve/pii/S2352302623001187