

Interview réalisée par
Clarisse GIRAULT
& Lydia CHERFAOUI
Internes en Hématologie
CHU de Nice
La myélofibrose est une néoplasie myéloproliférative chronique primitive ou secondaire caractérisée par une prolifération clonale myéloïde, associée dans 90 % des cas à des mutations dites « drivers » des gènes JAK2, CALR ou MPL, conduisant à une activation constitutive de la voie JAK-STAT, ainsi qu'à une fibrose médullaire progressive et une hématopoïèse extramédullaire.
L'expression clinique est hétérogène, et le risque est l'évolution vers une phase blastique. Malgré les progrès majeurs dans la compréhension des mécanismes physiopathologiques et l'intégration des données moléculaires dans les scores pronostiques récents, les options thérapeutiques approuvées demeurent limitées. Les inhibiteurs de JAK constituent le socle du traitement des formes symptomatiques, tandis que l'allogreffe de cellules souches hématopoïétiques reste la seule option curative chez les patients à haut risque.
Le Dr LOSCHI, maître de conférences en hématologie au CHU de Nice et expert de cette pathologie et membre du France Intergroup des syndromes myéloprolifératifs (FIM), apporte ici son éclairage sur le diagnostic, la stratification pronostique et la prise en charge thérapeutique actuelle de la myélofibrose.
Quels éléments clinico-biologiques doivent faire évoquer une myélofibrose ?
Il existe 2 tableaux clinico-biologiques qui doivent faire évoquer une myélofibrose :
- La forme proliférative (ou tumorale) qui se caractérise par une splénomégalie parfois volumineuse, associée à des degrés divers à des douleurs osseuses, de la fièvre, des sueurs nocturnes et une altération de l'état général.
L'hémogramme montre fréquemment une hyperleucocytose, une myélémie, la présence de blastes et d'érythroblastes circulants. - La forme cytopénique, plus insidieuse, peut se révéler par un syndrome anémique et/ou hémorragique. Elle peut être moins bruyante cliniquement ou associée à une volumineuse splénomégalie, dans ce cas en partie responsable des cytopénies. L'hémogramme met alors en évidence une ou plusieurs cytopénies.
Ces éléments clinico-biologiques sont d'autant plus évocateurs chez un patient suivi pour une polyglobulie primitive ou une thrombocytémie essentielle.
Quels critères permettent alors de poser le diagnostic ?
On distingue deux entités : la myélofibrose primitive et la myélofibrose secondaire à un syndrome myéloprolifératif.
La myélofibrose primitive est définie selon les critères OMS 2022. Le diagnostic nécessite la présence de 3 critères majeurs et au moins 1 critère mineur. Les critères majeurs sont :
- Histologique : fibrose médullaire de grade supérieure ou égale à 2 à la biopsie ostéo- médullaire, associée à une prolifération mégacaryocytaire atypique.
- Clonal : présence d'une mutation « driver » (JAK2, MPL et CALR) ou d'un autre marqueur clonal.
- Exclusion : absence d'arguments en faveur d'une autre hémopathie myéloïde ou d'une cause secondaire de fibrose médullaire (maladies auto-immunes, infections, autres néoplasies, lymphoproliférations, syndromes myélodysplasiques avec fibrose, etc.).
Les critères mineurs regroupent la présence d'érythroblastes circulants, une augmentation des LDH, une splénomégalie, une hyperleucocytose supérieure à 11 G/L et/ou une anémie non expliquée.
La myélofibrose secondaire correspond à l'évolution d'un syndrome myéloprolifératif préexistant, principalement une polyglobulie de Vaquez ou une thrombocytémie essentielle. Le diagnostic repose sur :
- L'existence documentée d'un syndrome myéloprolifératif initial.
- Et la mise en évidence, à la biopsie ostéo-médullaire, d'une progression vers une fibrose médullaire de grade supérieure ou égale à 2.
Chez un patient suspect de myélofibrose, la réalisation d'une biopsie ostéomédullaire est donc indispensable. La plupart des centres français experts dans la prise en charge de cette pathologie ont développé des stratégies pour aider à rendre cet examen moins douloureux :
- Protoxyde d'azote.
- Réalité virtuelle.
- Réalisation en radiologie interventionnelle sous anesthésie générale courte et contrôle scanographique.
L'analyse histologique est indispensable pour évaluer le degré de fibrose et quantifier l'infiltration blastique.
Quelle place occupe la caractérisation moléculaire par Next Generation Sequencing ?
Le séquençage par NGS myéloïde est recommandé lorsqu'un diagnostic de myélofibrose a été porté chez un patient dont l'état général est compatible avec une allogreffe ou un essai clinique (l'âge n'est pas une limite en soi). Il a un intérêt diagnostique avec l'identification d'un marqueur de clonalité (notamment des mutations « driver » JAK2, CALR et MPL), mais aussi pronostique avec la présence de la mutation CALR de type 1 (ou type 1-like) qui est de meilleur pronostic.
Le NGS permet également d'identifier des mutations additionnelles dites « à haut risque moléculaire » (ASXL1, SRSF2, EZH2, IDH1/2, U2AF1, TP53), aujourd'hui intégrées dans les scores pronostiques (sauf TP53 qui n'a pas encore été intégrée dans les scores mais confère un pronostic défavorable) et déterminantes dans la discussion d'une allogreffe de cellules souches hématopoïétiques (allo-HCT).
Dans le contexte d'allo-HCT, l'évaluation de la maladie résiduelle par NGS sur les mutations driver a montré un intérêt pronostique. Les travaux de Gagelmann et al. ont suggéré qu'une MRD négative précoce après allo-HCT (notamment à J30 et J90) est associée à un risque plus faible de rechute [1]. Des études françaises, notamment portées par la Société Francophone de Greffe de Moelle et de Thérapie Cellulaire (SFGM-TC), sont en cours pour mieux préciser l'impact pronostique de la persistance des mutations driver et des mutations additionnelles après allogreffe.
Quels sont les scores diagnostiques et pronostiques couramment utilisés ?
Les scores utilisés dans la myélofibrose ont pour but de stratifier les patients afin de guider la stratégie thérapeutique.
Pour la myélofibrose primitive, il est recommandé d'utiliser le MIPSS ou le MIPSS70+ version 2.0 qui intègre des critères clinico-biologiques (âge, altération de l'état général, cytopénie, dépendance transfusionnelle, hyperleucocytose, blastose circulante), cytogénétiques, moléculaires (ASXL1, EZH2, SRSF2, IDH) et le statut CALR. Il faut essayer d'utiliser ces scores les plus récents (MIPSS et MIPSS70+ v2.0) puisque les recommandations internationales de traitement, notamment les recommandations NCCN, se basent sur ces derniers et qu'une étude française a montré que ces scores permettent de porter plus précisément une indication d'allo-HCT [2].
Pour la myélofibrose secondaire, il est recommandé d'utiliser le MYSEC- PM basé sur des paramètres cliniques et biologiques (une version révisée de ce score intégrant elle aussi la biologie moléculaire est en cours de création).
Quelles sont les indications des traitements, et comment gérer leurs principales toxicités ?
En se basant sur les recommandations NCCN et britanniques [3, 4] si le patient est asymptomatique et qu'il a un score « très bas risque », une surveillance simple peut être proposée.
Dès que le patient commence à être symptomatique : splénomégalie volumineuse ou douloureuse, sueurs, perte de poids, douleurs osseuses, fièvre, un traitement est indiqué.
Il est important que pour chaque patient, chaque ligne thérapeutique soit autant que possible réalisée dans le cadre d'un essai clinique.
La première ligne de traitement repose sur les inhibiteurs de JAK (JAKi). Deux JAKi sont aujourd'hui approuvés en première ligne en France : le ruxolitinib et le fédratinib.
Leur efficacité est généralement évaluée au bout de 6 mois de traitement, notamment grâce au score RR6 (Response to Ruxolitinib at 6 months) pour les patients recevant une première ligne par ruxolitinib [5].
Le traitement de deuxième ligne en cas d'échec ou d'intolérance se fera préférentiellement dans le cadre d'un essai clinique.
Pour un patient présentant une récidive de symptômes ou une intolérance au ruxolitinib, en l'absence d'inclusion possible dans un essai clinique, le traitement recommandé est plutôt l'introduction d'un autre JAKi.
Une des principales causes d'intolérance au ruxolitinib est l'anémie. Il existe une corrélation directe entre cytopénies et la dose de ruxolitinib. Plus la dose de ruxolitinib est élevée, meilleure est l'efficacité sur les symptômes, mais plus le risque de cytopénies induites augmente.
Chez les patients traités par ruxolitinib en première ligne, le développement initial d'une anémie qui s'améliore à partir de la douzième semaine est classique et ne doit dans la majorité des cas, pas conduire à une modification thérapeutique.
Gestion des cytopénies chez les patients suivis pour myélofibrose : L'anémie est fréquente chez les patients suivis pour myélofibrose. Elle est multifactorielle, liée à la maladie, à la splénomégalie, à la dénutrition, à la toxicité des thérapeutiques.
Un tiers des patients sont anémiques au diagnostic et la quasi-totalité des patients deviennent anémiques au cours du suivi. Environ 50 % des patients seront transfusés au cours de leur première année de suivi.
Des études récentes retrouvent un impact direct de l'anémie sur la survie. De même, il existe une corrélation entre la dépendance transfusionnelle et la survie.

La gestion de l'anémie est décrite dans les récentes recommandations internationales. Pour les patients ne pouvant être inclus dans un essai clinique (notamment inhibiteurs de la voie du TGF-b). L'anémie classique survenant au cours des douze premières semaines de traitement par ruxolitinib ne doit pas forcément entraîner une modification thérapeutique. Au-delà, la persistance ou l'apparition d'une anémie doit faire envisager une modification thérapeutique.
Avant tout changement thérapeutique, une réévaluation médullaire et oncogénétique doit être réalisée afin de ne pas méconnaître une évolution clonale. En l'absence d'inclusion dans un essai clinique, deux stratégies sont possibles et proposées dans les recommandations britanniques.
La première stratégie consiste à remplacer le ruxolitinib par le momelotinib. Le momelotinib (inhibiteur de JAK et d'ACVR1) est autorisé en deuxième ligne en France sur la base de l'étude de phase III MOMENTUM. Il permet une amélioration de l'érythropoïèse, avec environ 40 % des patients transfusés qui deviennent indépendants des transfusions. Il est globalement bien toléré, malgré un risque de neuropathie et d'insuffisance rénale aiguë retrouvé dans les séries de vie réelles [6].
La deuxième stratégie envisagée dans les recommandations consiste à combiner une diminution de la posologie de l'anti-JAK pour diminuer la toxicité hématologique en y associant de l'EPO chez les patients présentant une EPO sérique inférieure à 500UI/L mais ceci peut s'accompagner d'une diminution de l'efficacité thérapeutique sur les symptômes, notamment la splénomégalie.
Si le patient présente une thrombopénie, la gestion est moins codifiée, mais les recommandations britanniques conseillent plutôt de changer d'inhibiteur de JAK. Si la thrombopénie apparaît sous ruxolitinib, il peut être intéressant d'essayer le fédratinib, ou le momelotinib, qui sont moins toxiques sur les plaquettes.
La transformation de la myélofibrose en phase accélérée ou blastique est également une indication à un changement de traitement.
Quand proposer une allogreffe de CSH ?
La proposition de l'allogreffe dépend avant tout de la stratification pronostique.
Pour les patients avec une myélofibrose primitive, une indication d'allogreffe est portée chez un patient présentant un score MIPSS/MIPSS70+v2.0 de haut risque ou très haut risque, même si dans certains cas (mutation TP53, âge), la possibilité d'une allogreffe peut être envisagée chez un patient présentant un risque intermédiaire. Pour les patients avec une myélofibrose secondaire à un autre syndrome myéloprolifératif, une indication d'allogreffe existe pour les patients présentant un score MYSEC-PM de haut risque.
L'indication d'allogreffe doit être réévaluée au cours de l'évolution en cas d'apparition de critères pouvant majorer les résultats des scores dynamiques MIPSS/MIPSS70+ v2.0.
Il est essentiel de discuter la greffe de CSH avant la transformation en phase accélérée ou blastique, car les résultats sont nettement moins favorables dans ce contexte. Tout patient présentant une indication d'allogreffe basée sur les scores précédemment cités doit bénéficier d'une évaluation par un hématologue greffeur.
Malgré une amélioration de la survie des patients allogreffés pour myélofibrose au cours des 10 dernières années, la principale cause de mortalité de la myélofibrose au cours de l'allogreffe de CSH est la toxicité de la procédure (maladie du greffon contre l'hôte, infections, défaillance d'organes, dysfonction du greffon). Prédire cette toxicité est donc essentielle et se base sur le calcul du score MTSS. La gestion de la splénomégalie avant l'allo greffier a fait l'objet de recommandations européennes [7]. De nouvelles stratégies sont en cours d'évaluation pour essayer de diminuer la toxicité et améliorer la prise du greffon.
Quels outils utiliser pour le suivi des patients et à quel rythme ?
Le suivi repose sur une approche multimodale clinique, biologique, histologique et radiologique. En pratique, les patients sont revus tous les trois à six mois. Les symptômes doivent être évalués à l'aide du MFSAF, associés à un examen clinique rigoureux, incluant la mesure objective de la taille de la rate en centimètres sous le rebord costal (et non sur les « travers de doigts »).
Le score RR6 [5] peut être calculé après six mois de traitement afin d'évaluer la réponse thérapeutique.
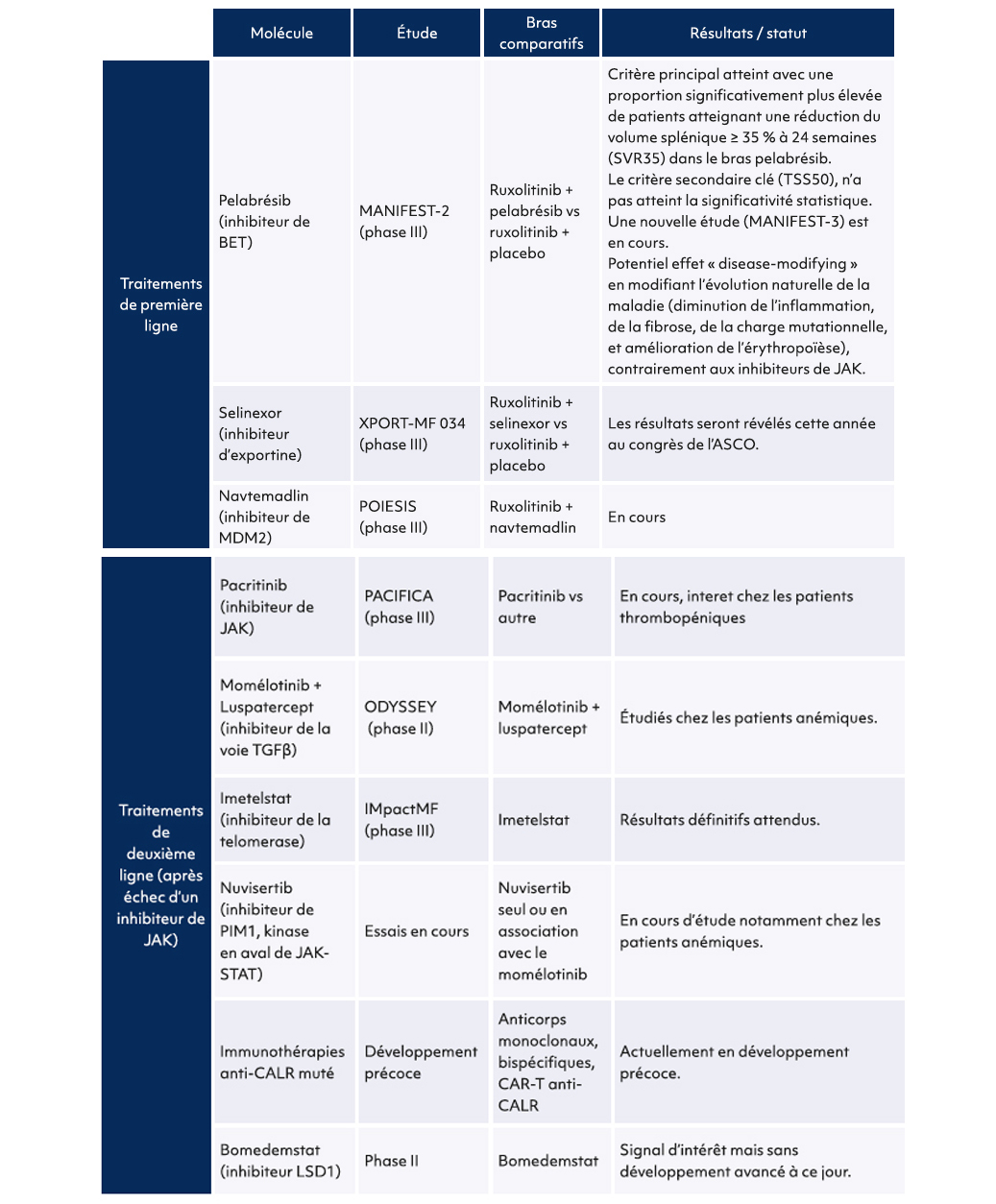
Quelles sont les nouvelles perspectives thérapeutiques ?
Il existe de nombreux développements dans la myélofibrose :
Quels conseils donneriez-vous à un interne face à un cas de myélofibrose ?
- La confirmation histologique du diagnostic par biopsie ostéomédullaire est indispensable.
- Le NGS myéloïde est essentiel. Il a une valeur diagnostique (clonalité), pronostique (mutations de haut risque) et thérapeutique (stratification). Il est également essentiel en post-allo-HCT pour le suivi de la maladie résiduelle.
- Il faut renouveler la BOM et le NGS, en cas d'évolution d'un syndrome myéloprolifératif vers une myélofibrose, de rechute, ou de transformation en phase accélérée.
- Adresser précocement les patients à un expert de la pathologie pour évaluation : Tout patient doit pouvoir bénéficier d'une évaluation en centre régional spécialisé, notamment pour discuter de l'inclusion dans un essai clinique, et évaluer l'indication d'une allogreffe de CSH.
- Ne pas oublier la prophylaxie anti- infectieuse sous inhibiteurs de JAK. Une prophylaxie contre la réactivation du VZV doit être envisagée. La vaccination par Shingrix est recommandée chez les patients éligibles ; en cas d'antécédent de zona, une prophylaxie antivirale par valaciclovir peut être proposée.
- Concernant l'allo-HCT : le plus tôt est le mieux pour les patients ayant une indication définie par les scores cités plus haut.
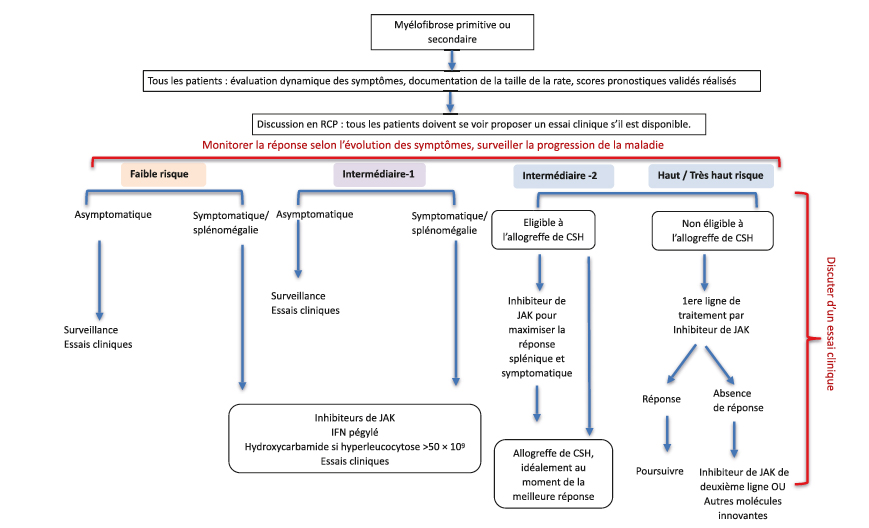
Figure 1 : Recommandations britanniques de la prise en charge thérapeutique de la Myélofibrose.
Bibliographie
1. Gagelmann N, Kröger N. Allogeneic Stem Cell Transplant for Myelofibrosis and Myelodysplastic Syndromes: A Contemporary Review. Am J Hematol. 2025 Jun;100(Suppl 4):16–29.
2. Luque Paz D, Gagelmann N, Benajiba L, Riou J, Salit R, Orvain C, et al. Role of molecular alterations in transplantation decisions for patients with primary myelofi brosis. Blood Adv. 2024 Nov 23;9(4):797–807.
3. mpn.pdf [Internet]. [cited 2026 Feb 6]. Available from: https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf
4. McLornan DP, Psaila B, Ewing J, Innes A, Arami S, Brady J, et al. The management of myelofi brosis: A British Society for Haematology Guideline. British Journal of Haematology. 2024;204(1):136–50.
5. Maffioli M, Mora B, Ball S, Iurlo A, Elli EM, Finazzi MC, et al. A prognostic model to predict survival after 6 months of ruxolitinib in patients with myelofibrosis. Blood Adv. 2022 Mar 17;6(6):1855–64.
6. Pérez-Lamas L, Segura Diaz A, García Delgado R, Álvarez-Larrán A, Senin MA, Mora E, et al. Real world outcomes of momelotinib in myelofibrosis patients with anemia: results from the MOMGEMFIN study. Blood Cancer J. 2025 Apr 17;15(1):67.
7. Polverelli N, Hernández-Boluda JC, Czerw T, Barbui T, D'Adda M, Deeg HJ, et al. Splenomegaly in patients with primary or secondary myelofi brosis who are candidates for allogeneic hematopoietic cell transplantation: a Position Paper on behalf of the Chronic Malignancies Working Party of the EBMT. Lancet Haematol. 2023 Jan;10(1):e59–70.

Dr Michael LOSCHI
Maitre de conférences
en hématologie
CHU de Nice