


Pierre-Edouard Debureaux1, Olivier Lidove2, Pierre Lemaire3, Anne-Marie Zagdanski4, Wladimir Mauhin2, Agathe Grandjean5, Stéphanie Harel1, Alexis Talbot1, Bertrand Arnulf1 et Bruno Royer1
1 Service d'Immuno-Hématologie, Hôpital Saint Louis, Paris, France
2 Service de Médecine Interne, Centre de Référence Maladies Lysosomales, Groupe Hospitalier Diaconesses-Croix Saint-Simon, Paris, France
3 Service d'Hématologie Biologique, Hôpital Saint Louis, Paris, France
4 Service d'Imagerie, Hôpital Saint Louis, Paris, France
5 Service de Rhumatologie, Groupe Hospitalier Diaconesses-Croix Saint-Simon, Paris, France
Nous rapportons le cas d'une patiente âgée de 56 ans, adressée en immuno-hématologie à l'hôpital Saint Louis pour l'exploration d'un pic monoclonal.
La patiente est suivie depuis l'enfance pour une maladie de Niemann- Pick de type B. À l'âge de 10 ans par une baisse de l'activité de la sphingomyélinase acide (activité 0.18 μkat/kg protéine, donc activité résiduelle notable confirmée sur fibroblastes retrouvant un profond déficit, avec activité "in situ" sur cellules vivantes notable) et une mutation homozygote R610del du gène SMPD1 ont permis le diagnostic de Niemann-Pick de type B. La patiente a été suivie en centre de référence. Elle présente comme signes de cette pathologie chronique une hépatosplénomégalie, un syndrome interstitiel pulmonaire entraînant un trouble ventilatoire mixte, une anémie (hémoglobine 11,8 g/dL) et thrombopénie modérées (Plaquettes 100 G/L), une dyslipidémie, ainsi qu'une ostéoporose. Elle ne reçoit pas de traitement substitutif enzymatique.
Dix ans avant la consultation en hématologie, l'hémogramme montrait une hémoglobine à 12 g/dL, des plaquettes à 94 G/L, pas de pic monoclonal détecté., L'imagerie de l'époque retrouve une hépatomégalie avec une flèche à 21 cm et splénomégalie à 12,8 cm. Un an avant la consultation, un bilan systématique révèle un pic monoclonal IgA kappa dont le taux est mesuré à 9,4 g/L, sans critère CRAB (Hb 11,8 g/dL, plaq 100 G/L, créatinine 49 μmol/L), avec une hépatosplénomégalie stable. Dans les mois précédent la consultation en hématologie, le pic a augmenté jusqu'à 11,1 g/L, l'anémie s'aggrave (hémoglobine à 10,9 g/dL) et des fractures vertébrales (T10, T12) sont constatées, décrites comme ostéoporotiques.
Lors de la consultation en hématologie à 56 ans, un bilan à la recherche des critères CRAB-SLIM (1) est réalisé : calcémie corrigée à 2,44 mmol/L, créatinine 50 μmol/L, protéinurie inférieur 0,5 g/g, anémie à 10,4 g/dL, thrombopénie stable à 90 G/L, fractures vertébrales ostéoporotiques thoraciques et lombaires, un myélogramme montrant 50 % de plasmocytes dystrophiques et des macrophages en « bleu-de-mer » (caractéristique de sa maladie de surcharge, Figure 1), un ratio chaînes légères sériques à 10, et pas de lésions rattachées au myélome à l'IRM (fractures d'allure ostéoporotique).
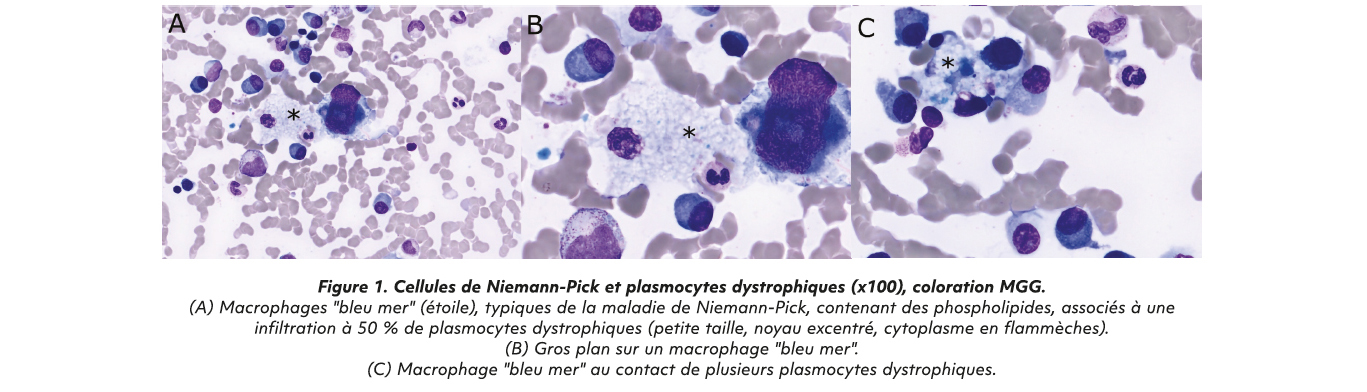
Au total, le diagnostic de myélome asymptomatique a été retenu mais au vu de la progression de l'anémie et des lésions osseuses, un suivi rapproché associé à une relecture des imageries retrouvent une majoration de l'anémie à 9,8 g/dL et la présence de lésions focales supra-centimétrique à l'IRM associés aux fractures ostéoporotiques (Figure 2) qui ont mené à l'initiation d'un traitement. Il n'a pas été décelé de translocation t(11;14) ou de délétion 17p en FISH sur plasmocytes.
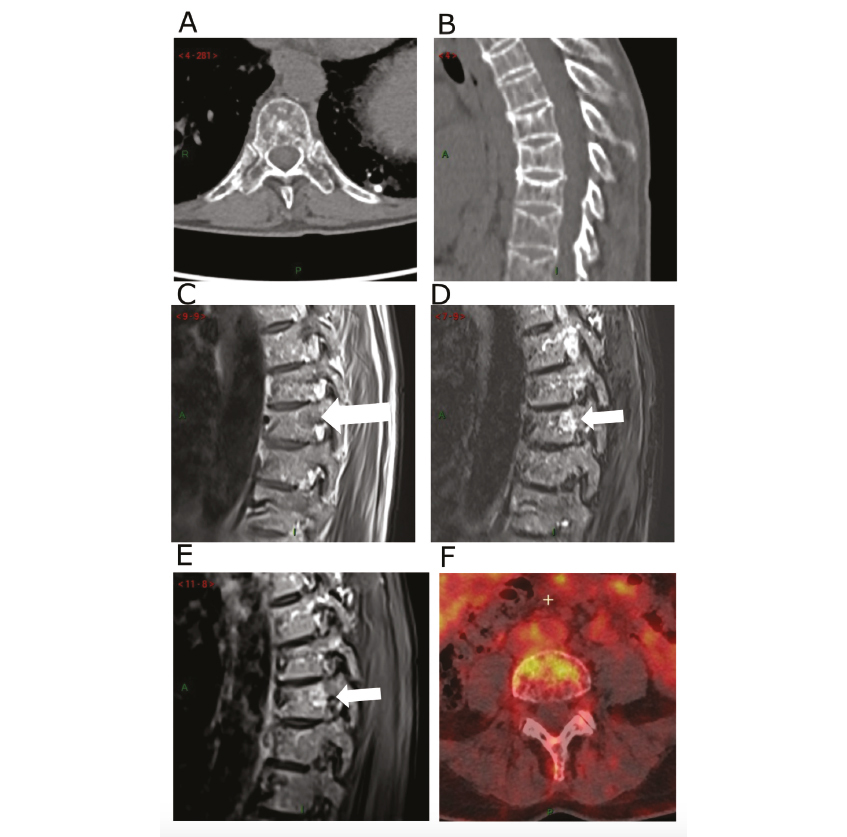
Figure 2. Bilan d'imagerie au diagnostic du myélome
(A-B) Scanner low dose non injecté du rachis thoraco-lombaire retrouvant une déminéralisation avec une trame osseuse hétérogène micro lacunaire au niveau lombaire (A) et des tassements vertébraux thoraciques (B) par insuffisance osseuse dans le cadre de l'ostéoporose. (C-E) IRM du rachis thoracique retrouvant une lésion focale de 8mm au niveau T8 (flèche) avec un hyposignal T1(C), un hypersignal après injection de gadolinium (D) et en séquence STIR (E). (F) TEP-scanner au 18-FDG retrouvant une fixation hypermétabolique avec SUV 4,6 d'une lésion du rachis lombaire.
En RCP, un traitement intensif est validé par quatre cycles de daratumumab- bortezomib-lénalidomide- dexaméthasone (DVRD), suivi d'une autogreff e conditionnée par melphalan forte dose à 200 mg/m², de deux cures de consolidation DVRD, et de deux ans de maintenance par lénalidomide 5 mg en continu. Un traitement par denosumab à dose oncologique (120 mg par mois) est instauré. La tolérance de l'ensemble des traitements est bonne sans complication majeure. Lors de la dernière consultation de suivi à 2 ans et demi du début de la prise en charge, la patiente est en rémission immunochimique persistante pour le myélome, l'électrophorèse des protéines sérique est normale, les gammaglobulines polyclonales sont mesurées à 8,7 g/L, il persiste des cytopénies modérées (hémoglobine 12,9 g/dL et plaquettes 94 G/L), la fonction rénale est stable, il n'y a pas de protéinurie détectée, et les bilan phospho-calcique et hépatique sont normaux. Il n'a pas été débuté de traitement substitutif pour la maladie de Niemann-Pick durant le traitement du myélome et elle est sous denosumab à dose rhumatologique. Lors de la dernière RCP de sa maladie de surcharge, il a été décidé de débuter prochainement un traitement de supplémentation enzymatique à la fin du traitement d'entretien du myélome.
Discussion
Le déficit en sphingomyélinase acide, également appelée maladie de Niemann-Pick, est une maladie génétique de transmission autosomique récessive rare liée à des mutations du gène SMPD1 (2). Il s'agit d'une maladie de surcharge lysosomale (comme la maladie de Gaucher), à l'origine d'une accumulation ubiquitaire en sphingomyéline (un composant des membranes cellulaires) et d'autres lipides au cours de la vie, à l'origine de manifestations cliniques systémiques. Cette accumulation affecte principalement les cellules du système réticulo-endothélial, ce qui explique les manifestations principales, que sont l'hépatosplénomégalie et l'atteinte parenchymateuse pulmonaire. Le myélogramme quand il est réalisé peut mettre en évidence une cellule caractéristique, le macrophage (précédemment dénommé histiocyte) « bleu de mer ». La patiente présente ces signes cliniques au sein de la forme viscérale de l'adulte de type B. Des douleurs osseuses et de l'ostéoporose ont été décrits chez 20 % des patients (3). Il existe un traitement substitutif pour les formes sans atteintes sévères du système nerveux (formes B et AB éligibles au traitement), l'olipudase alfa dont l'indication doit être validé en RCP spécialisée (4, 5). La première cause de décès chez les patients atteints Niemann-Pick du type B est le cancer (6).
Chez les patients suivi pour une maladie de Niemann-Pick de type B, un premier cas de myélome IgG lambda symptomatique a été rapporté par le Dr E. Portier dans la même équipe d'hématologie qui a décrit ce cas (7). Ce patient avait été initialement étiqueté maladie de Gaucher, il présentait les mêmes atteintes d'organes (hépatosplénomégalie et pulmonaire) et une infiltration plasmocytaire de 41 % associé aux macrophages « bleu de mer » (cellule non retrouvée dans la maladie de Gaucher) avec une issue favorable pour le moment. Notre patiente constitue ainsi le deuxième cas documenté en France, et il existe au moins un troisième cas documenté aux États-Unis (données personnelles). Les maladies de surcharge lysosomale, notamment Gaucher et Niemann-Pick, exposent à un risque accru de gammapathies monoclonales. Dans la maladie de Gaucher type 1, la prévalence de la phase pré-myélome qu'est la gammapathie monoclonale de signification indéterminée (MGUS en anglais) atteint 32 %, et le risque de myélome est particulièrement élevé par rapport à la population générale (RRx25-51) (8). Dans une série française de suivi des patients avec Niemann-Pick de type B menée par le Dr Lidove, 6 patients sur 28 présentaient une hypergammaglobulinémie dont 5 MGUS mais aucun cas de myélome n'avait été décrit à l'époque (3).
En termes de physiopathologie, une des hypothèses pour expliquer cette prévalence élevée de gammapathie dans la maladie de Gaucher serait liée à une stimulation immune des plasmocytes par l'accumulation des deux lysolipides qui générerait des anticorps dirigés contre eux (9). Le blocage de la synthèse de ces 2 lysolipides chez la souris avec un équivalent de maladie de Gaucher a limité l'apparition d'hémopathie B (10). La même hypothèse est émise pour cette association d'une maladie de Niemann-Pick et d'un myélome multiple (pas de données physiologiques disponibles). Nous pouvons nous poser la question de l'indication d'un traitement substitutif en cas d'apparition de MGUS chez ces patients.
En présence d'un myélome multiple et des fractures ostéoporotiques, l'utilisation de dénosumab s'est imposée : les bisphosphonates sont évités en raison de l'eff et inhibiteur de la sphingomyélinase (11).
Enfin, ce cas met en évidence l'importance d'une relecture experte des examens d'imagerie afin de distinguer les lésions ostéolytiques liées au myélome des modifications osseuses secondaires à la maladie de surcharge.
Bien que rare, l'association entre maladie de surcharge lysosomale et myélome justifie un dépistage systématique des anomalies monoclonales chez ces patients, notamment en cas d'antécédent de MGUS ou de signes cliniques évocateurs. Il faudra également rechercher des formes frustes de ces maladies de surcharge en cas d'hépatosplénomégalie chez des patients au diagnostic de myélome via un dosage des enzymes puis une confirmation génétique. Il faudra également à l'esprit l'hypothèse d'une amylose AL qui peut aussi donner une splénomégalie au cours du myélome (association bien plus fréquente).
Référence
1. Vincent L, Roussel M, Macro M, Karlin L, Vekemans MC, Royer B, et al. Recommandations 2024 de l'Intergroupe francophone du myélome sur la prise en charge des gammapathies monoclonales de signification indéterminée: Hématologie. 1 janv 2024;30(1):71-88.
2. Michaud M, Mauhin W, Villeneuve T, Lidove O. [Acid sphingomyelinase deficiency: A review]. Rev Med Interne. 24 mai 2025;S0248- 8663(25)00506-5.
3. Lidove O, Belmatoug N, Froissart R, Lavigne C, Durieu I, Mazodier K, et al. Déficit en sphingomyélinase acide (maladie de Niemann-Pick B) : une étude rétrospective multicentrique de 28 patients adultes. Rev Médecine Interne. mai 2017;38(5):291-9.
4. Diaz GA, Jones SA, Scarpa M, Mengel KE, Giugliani R, Guff on N, et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genet Med. août 2021;23(8):1543-50.
5. Wasserstein M, Lachmann R, Hollak C, Arash-Kaps L, Barbato A, Gallagher RC, et al. A randomized, placebo-controlled clinical trial evaluating olipudase alfa enzyme replacement therapy for chronic acid sphingomyelinase deficiency (ASMD) in adults: One-year results. Genet Med. juill 2022;24(7):1425-36.
6. Mauhin W, Guff on N, Vanier MT, Froissart R, Cano A, Douillard C, et al. Acid sphingomyelinase deficiency in France: a retrospective survival study. Orphanet J Rare Dis. 5 août 2024;19(1):289.
7. Portier E, Talbot A, Nguyen Y, Royer B, Pettazzoni M, Ben Salah I, et al. Multiple myeloma occurring in a case of Niemann‐ Pick disease Type B: A pathophysiological link? Br J Haematol [Internet]. mai 2022 [cité 11 juin 2025];197(4). Disponible sur : https://onlinelibrary.wiley.com/doi/10.1111/bjh.18050
8. Arends M, van Dussen L, Biegstraaten M, Hollak CEM. Malignancies and monoclonal gammopathy in Gaucher disease; a systematic review of the literature. Br J Haematol. juin 2013;161(6):832-42.
9. Nair S, Sng J, Boddupalli CS, Seckinger A, Chesi M, Fulciniti M, et al. Antigen-mediated regulation in monoclonal gammopathies and myeloma. JCI Insight [Internet]. 19 avr 2018 [cité 11 juin 2025];3(8). Disponible sur : https://insight.jci.org/articles/view/98259
10. Pavlova EV, Archer J, Wang S, Dekker N, Aerts JM, Karlsson S, et al. Inhibition of UDP-glucosylceramide synthase in mice prevents Gaucher disease-associated B-cell malignancy. J Pathol. janv 2015;235(1):113-24.
11. Roth AG, Drescher D, Yang Y, Redmer S, Uhlig S, Arenz C. Potent and Selective Inhibition of Acid Sphingomyelinase by Bisphosphonates. Angew Chem Int Ed. 2009;48(41):7560-3.

Dr Pierre-Edouard DEBUREAUX
Hématologue à l'Hôpital Saint-Louis