

Parmi les hémopathies à cellules dendritiques, la classification OMS 2022 permet la distinction entre la leucémie à cellules dendritiques plasmocytoïdes (LpDC) et les proliférations matures de cellules dendritiques associées à une hémopathie (MPDCP). La LpDC est une hémopathie rare du sujet âgé dont les particularités de sa présentation clinique et biologique en font une entité de diagnostic souvent difficile pour les hématologues et pour les biologistes. Le pronostic est défavorable avec des rechutes retrouvées chez la majorité des patients, et ce, malgré une amélioration du pronostic en cas de recours à la greffe de cellules souches hématopoïétiques. Ces aspects en font une hémopathie dont le diagnostic doit être précoce et précis afin de proposer la prise en charge la plus adaptée aux patients.
Une hémopathie développée à partir de cellules du système immunitaire
La leucémie à cellules dendritiques plasmocytoïdes (LpDC) ou Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) trouve sa contrepartie physiologique dans les cellules dendritiques plasmocytoïdes (pDC). Par opposition aux cellules dendritiques
conventionnelles (cDC), les pDC sont des cellules du système immunitaire innée dont le principal rôle est la sécrétion d'interféron de type I en réponse à une stimulation immunitaire, le plus souvent d'origine virale1 . Ces cellules sont produites dans la moelle osseuse et vont se localiser principalement dans les ganglions lymphatiques ou le tissu lymphoïde associé aux muqueuses (MALT) dans divers sites anatomiques (tube digestif, peau…)2 .
Épidémiologie
La LpDC est rare avec une incidence globale estimée à 0,5 cas/ million habitants/an ce qui représente entre 35 et 40 nouveaux cas diagnostiqués en France chaque année. Elle touche de façon prédominante le sujet âgé, avec un âge médian au diagnostic de 68 ans et un sexe-ratio déséquilibré de 4 hommes pour 1 femme3, 4. Cette distribution est cependant plutôt bimodale avec un premier pic d'incidence retrouvé avant 20 ans. Les cas pédiatriques sont donc rares mais existent avec des découvertes possibles dès la première année de vie 5.
Présentation clinique
Les symptômes cutanés sont les manifestations les plus fréquentes dans la LpDC avec un pourcentage de patient atteint d'environ 70 à 90 % au diagnostic3, 4, 6. Ces lésions cutanées sont variées dans leur présentation et vont concerner principalement les territoires photo-exposés. Elles peuvent représenter la seule atteinte de la maladie (jusqu'à 30 % des patients selon les séries).
Un syndrome tumoral est fréquemment retrouvé, en particulier sous la forme d'adénopathies (394 à 56 %3 des patients) ou d'une splénomégalie (22 % (6) à 44 %3) avec ou sans hépatomégalie (17 %6 à 42 %3).
L'atteinte du système nerveux central (SNC) est également décrite avec une prévalence précise difficilement évaluable. Ainsi dans une cohorte rétrospective américaine, cette atteinte était asymptomatique chez 57 % des 103 patients évalués. Il est également possible d'assister à des rechutes neuroméningées chez des patients sans atteinte neurologique initiale avec une rechute parfois décrite uniquement au niveau du SNC. L'atteinte initiale du SNC est associée à une fréquence plus importante de rechute, appuyant l'intérêt de la dépister au diagnostic de la maladie 7 .
Diagnostic biologique
Hémogramme
La présentation biologique de la LpDC est en lien avec la présence des cellules leucémiques dans les différents territoires anatomiques. Si les pDC matures physiologiques sont rares dans le sang, les cellules leucémiques de LpDC, aussi appelées « blastes pDC », sont retrouvées dans le sang périphérique d'environ 70 % des patients. Cette présence de blastes est parfois à l'origine d'une hyperleucocytose avec des taux de globules blancs pouvant dépasser les 50 G/L, mais les patients sont le plus souvent leucopéniques avec une leucocytose moyenne de 1 G/L3.
L'atteinte médullaire (62 %4 à 96 %3 des patients selon les cohortes) est à l'origine d'une thrombopénie fréquente (75 %) et d'une anémie chez la plupart des patients sans atteinte cutanée isolée.
Aspects cytologiques
En cas de dissémination de la pathologie, l'examen cytologique du frottis sanguin ou médullaire est la première étape de l'orientation diagnostique d'une LpDC. La morphologie typique est celle de cellules de taille moyenne présentant un noyau rond, le plus souvent régulier, souvent excentré et au nucléole visible. Le cytoplasme est légèrement basophile et contient des petites vacuoles disposées en collier de perles sous le noyau. La présence d'un pseudopode donne à la cellule sa forme classique en miroir à main. (Figure 1A)3.
Les aspects cytologiques sont cependant souvent trompeurs et le cytologiste, même averti, pourra évoquer un diagnostic de lymphome ou de leucémie aiguë plutôt que celui d'une LpDC (Figure 1B).
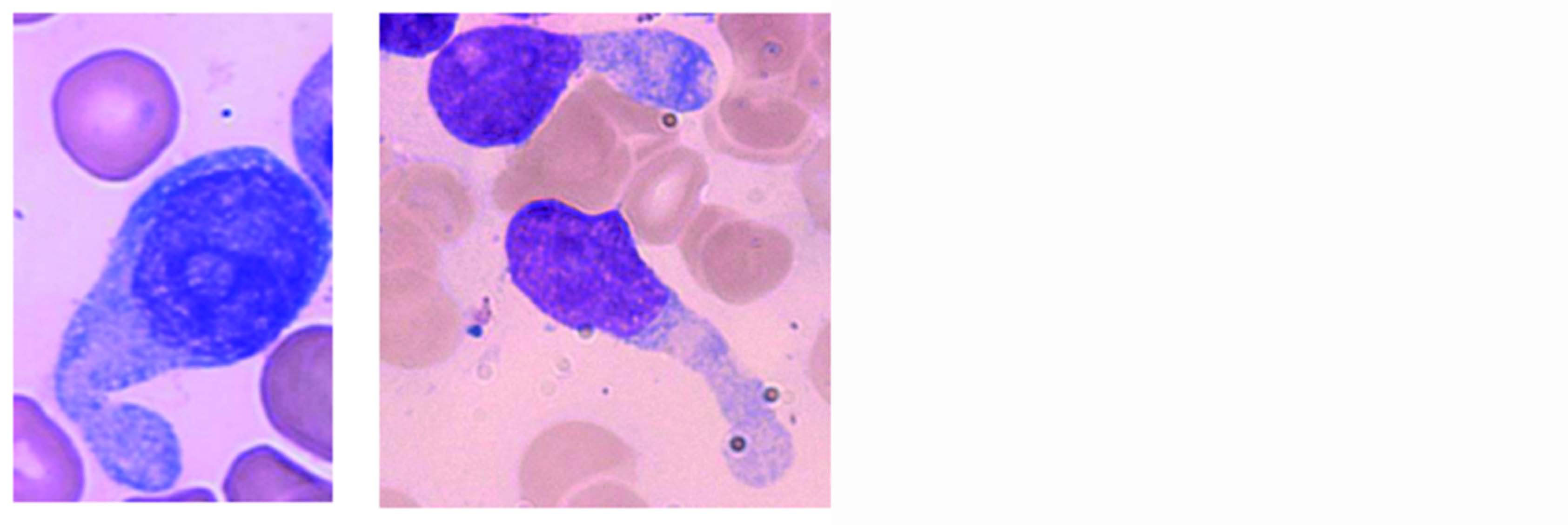
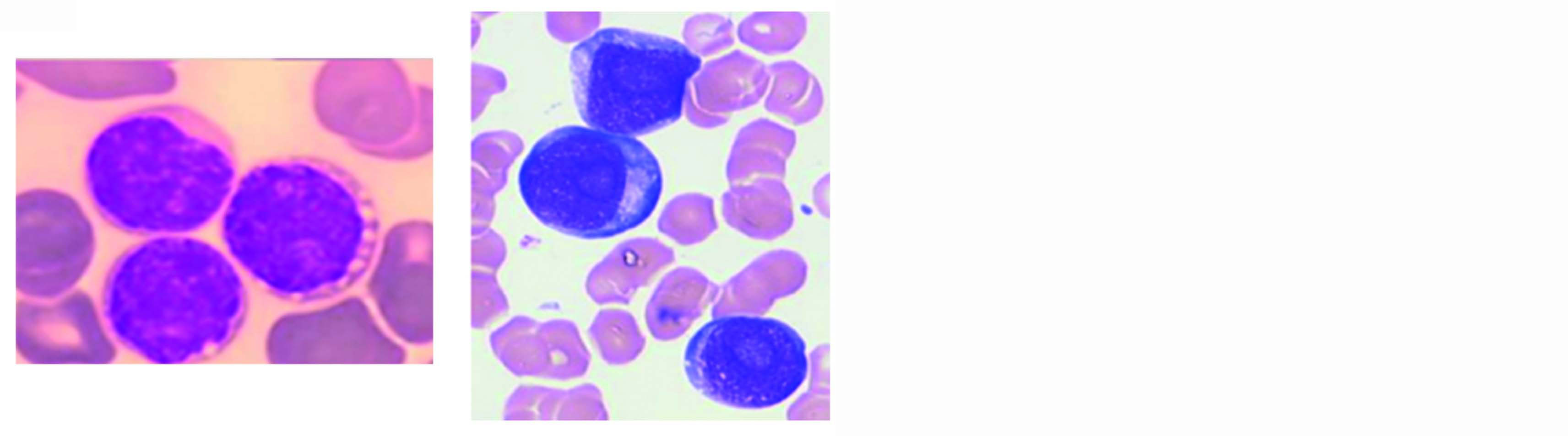
Figure 1 : Aspects cytologiques retrouvés dans la LpDC. (A) La morphologie classique est celle de cellules dites en "miroir à main". (B) Des présentations atypiques sont parfois retrouvées avec des aspects pseudo-lymphomateux ou monoblastiques. Crédits : Pr Francine GARNACHE-OTTOU.
Cytométrie en flux – pensez à « 1, 2, 3, 4, 5, 6 » !
En présence de cellules tumorales circulantes ou envahissant la moelle osseuse, la confirmation diagnostique nécessitera d'avoir recours à un immunophénotypage par cytométrie en flux (CMF). Celui-ci permettra de mettre en évidence des cellules de type pDC par l'expression des marqueurs de lignée classiques à savoir CD4, CD123, CD45RA, HLA-DR, FCeRI ainsi que CD303 (BDCA2) et CD304 (BDCA4), spécifiques de la lignée pDC, mais
dont l'intensité d'expression peut être diminuée voire négative. C'est la présence du marqueur pathologique CD56 sur la population suspecte qui permettra d'évoquer le diagnostic d'une LpDC (Figure 2). Les cellules leucémiques n'expriment jamais le marqueur d'immaturité CD34, et expriment parfois de façon aberrante des marqueurs des lignées lymphoïdes T (CD7 dans 40-60 % des cas, CD2 dans 30-40 % des cas), B (CD22 dans 20 % des cas) ou myéloïde (CD33 dans 45 % des cas). À noter l'absence d'expression des marqueurs de maturité monocytaire (CD14, CD11b ou encore CD64) à l'exception du CD36 également exprimé sur les pDC physiologiques3.
Des marquages intracytoplasmiques peuvent également être réalisés, à la recherche du marqueur TCL1, exprimé dans quasiment 100 % des cas, et également dérégulé dans la leucémie prolymphocytaire T8.
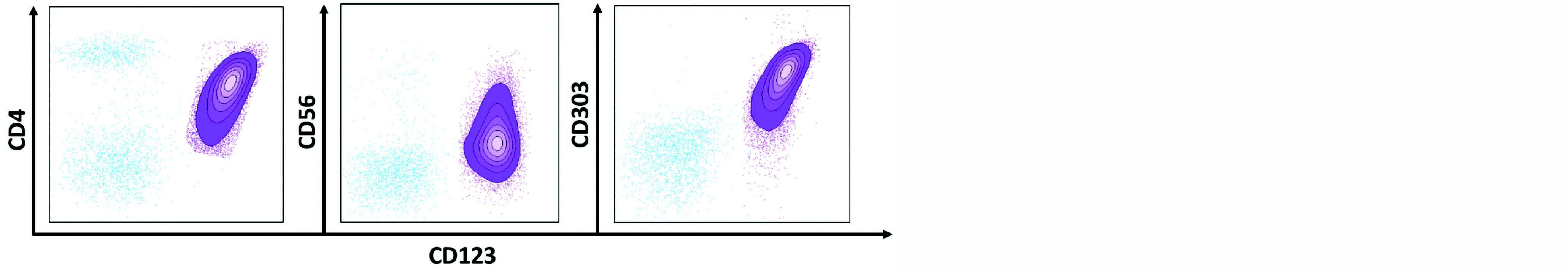
Figure 2 : Graphes de cytométrie typiques d'un cas de LpDC. Mise en évidence d'une population de cellules de type pDC (en violet) positives pour les marqueurs CD123, CD4 et CD303 exprimant de façon pathologique le CD56, en comparaison avec ces mêmes marqueurs sur les lymphocytes (en bleu).
Afin d'aider les biologistes pour le diagnostic, un algorithme d'analyse des marqueurs phénotypiques par CMF ainsi qu'un « score LpDC » ont été proposés en 2009 par Garnache-Ottou et al9. Ce score, sur 5 points, permet de distinguer la LpDC d'autres hémopathies malignes dont la présentation biologique et clinique peuvent être similaires. Un score supérieur ou égale 3 permet de confirmer le diagnostic de LpDC. (Tableau 1)
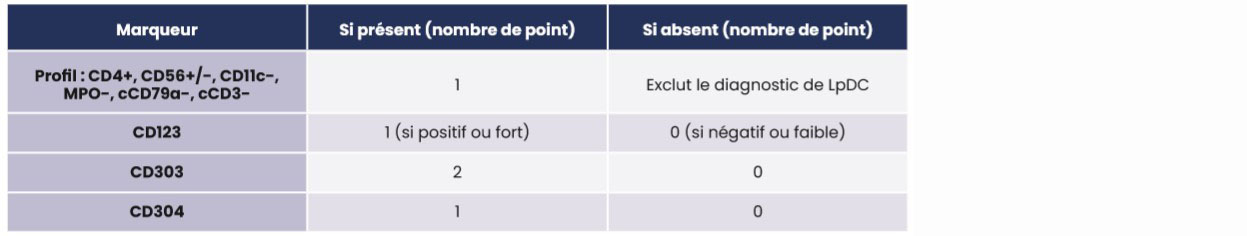
Tableau 1. Score « LpDC » tel que proposé par Garnache-Ottou et al9.
Le principal diagnostic différentiel sur les plans cytologique et cytométrique sera celui d'une leucémie aiguë associée à un excès de pDC (LAM-pDC). Cette entité, retrouvée dans la classification OMS 2022 au sein des MPDCP (Mature Plasmacytoid Dendritic Cell Proliferation), associe les critères de diagnostic d'une leucémie aiguë myéloblastique (LAM) selon les critères classiques et la prolifération de pDC de phénotype « physiologique » différent de celui de la LpDC. La cytologie est le plus souvent celle d'une LAM0 ou LAM5 de l'ancienne classification FAB et il est souvent difficile de différencier les blastes et les pDC.
En CMF, on identifie une population de blastes myéloïdes (CD34+ ou CD34- selon le type de LAM) et un contingent, plus ou moins important mais dont le seuil est dans la littérature fixé à au moins 2 %, de pDC CD123+ CD4+ mais CD56-10, 11.
Aspects moléculaires et cytogénétiques
Les anomalies moléculaires rencontrées dans la LpDC sont complexes puisqu'elles associent des anomalies rencontrées à la fois dans les hémopathies myéloïdes et lymphoïdes. Ce profil atypique traduit l'ontogénie complexe des pDC, dont l'origine myéloïde ou lymphoïde, bien qu'abondamment décrite dans les modèles murins, est encore la source de débats chez l'être humain à l'heure actuelle12, 13. Ainsi, dès son introduction dans la classification OMS 2008, la LpDC était classé au sein des hémopathies myéloïdes4. Ce n'est que dans la dernière édition de 2022 qu'elle a été individualisée au sein des hémopathies histiocytaires et des cellules dendritiques15.
La LpDC est ainsi caractérisée par des anomalies des régulateurs épigénétiques (TET2 en premier lieu ainsi qu'IDH1 et DNMT3A) et de l'épissage avec des anomalies des gènes des composants du spliceosome (SF3B1, SRSF2, U2AF1 et ZRSR2)16. En lien avec les anomalies fréquentes de TET2 (40 à 60 % des cas), des équipes proposent que les pDC ayant acquis ces mutations seraient plus résistantes à l'apoptose induite par l'exposition aux UV avec l'accumulation secondaire d'anomalies génétiques et le développement du processus tumoral, en premier lieu au niveau cutané, avec une potentielle dissémination dans le reste de l'organisme17.
Ces anomalies partagées entre la LpDC et les hémopathies myéloïdes expliquent peut-être leur association fréquente. En effet, on retrouve une hémopathie myéloïde concomitante, ou dans les antécédents du patient, dans environ 20 % des cas18, 19.
Parmi les anomalies lymphoïdes, on retrouve dans 70 % des cas environ une inactivation biallélique d'ETV620. On retrouve également des anomalies du gène IKZF16.
Les anomalies chromosomiques sont fréquentes dans la LpDC et concernent entre 57 et 75 % des patients au diagnostic avec un caryotype complexe retrouvé chez environ 50 % d'entre eux. Les régions chromosomiques les plus fréquemment dérégulées sont par ordre de fréquence : 5q (72 %), 12p (64 %), 6q (50 %), 15q (43 %) et le chromosome 9 (28 %)6.
Des anomalies cytogénétiques impliquant plus particulièrement l'oncogène MYC sont retrouvées dans environ 30 % des cas. Ces anomalies rapprochent la LpDC des lymphomes de haut grade. L'une des anomalies entraînant la dérégulation de cet oncogène est la translocation t(6;8) qui conduit à la juxtaposition au promoteur du gène RUNX2, impliqué dans la différenciation pDC. Cette anomalie, spécifique de la LpDC, pourrait contribuer à la pathogenèse de la maladie en activant le programme transcriptionnel pDC dans les cellules tumorales tout en entraînant une dérégulation du cycle cellulaire et une résistance à l'apoptose par l'activation des cibles de MYC19.
La LpDC est également caractérisée par une fréquence importante de réarrangements du gène MYB. En interaction étroite avec MYC, il est impliqué dans la survie et la prolifération cellulaires. Dans une cohorte de 14 cas, il a été retrouvé des réarrangements de MYB chez neuf patients dont 100 % des cas pédiatriques étudiés (cinq cas). Les réarrangements de MYB semblent mutuellement exclusifs des anomalies de MYC dans la LpDC et associés à un meilleur pronostic que ces dernières21.
Traitement et pronostic
Approche conventionnelle
Les modalités thérapeutiques de la LpDC se sont d'abord limitées à des approches de chimiothérapie conventionnelle, associées, ou non, à une greffe de cellules souches hématopoïétiques (CSH). Ils sont classiquement divisés en traitement de type leucémies aiguës « AL-type » ou ceux utilisés dans les lymphomes non hodgkiniens « NHL-type ».
Les données les plus complètes proviennent d'une cohorte internationale rétrospective composée de 398 patients atteints de LpDC suivis de 2001 à 20174. Tout traitement confondu, la survie globale (Overall Survival ou OS) à 5 ans était de 16,8 %. Ce faible taux de survie était surtout observé chez les patients n'ayant été traité que par un régime de chimiothérapie conventionnelle, AL ou NHL-like, avec une OS à 5 ans inférieure à 10 %. Ce pronostic défavorable était amélioré par le recours à une procédure de greffe de CSH puisque l'on observe une OS à 5 ans de 54 % dans le groupe « chimiothérapie + autogreffe » (16 patients) et de 56,2 % dans le groupe « chimiothérapie + allogreffe » (61 patients). Enfin, dans cette cohorte on observe une rechute chez 76 % des patients tout traitement confondu. Le plus important taux de rechute était observé chez les patients n'ayant pas reçu de greffe (78 % des patients analysés) avec un taux de rechute estimé à environ 30 % en cas d'allogreffe ou d'autogreffe de CSH.
Il existe peu de données permettant de comparer l'autogreffe à l'allogreffe et les résultats sont discordants entre les équipes. Ainsi, une équipe japonaise retrouve un effet favorable de l'autogreffe parmi 11 patients autogreffés et comparés à 14 patients allogreffés avec une survie à 4 ans supérieure dans le premier groupe (82 % contre 69 %)22.
À l'inverse, les données de la cohorte française publiées en 2019 sont favorables à l'allogreffe. Sur la période de 2000 à 2013, 78 patients étaient analysables sur le plan thérapeutique et 34 ont bénéficié d'une greffe de CSH, dont 4 autogreffes. Tous les patients du groupe autogreffe ont rechuté et sont décédés et des rechutes ont été observées chez 33 % des patients allogreffés3.
Il apparaît que le pronostic des patients est nettement amélioré lorsqu'une greffe de CSH est pratiquée. Les meilleures chances de survie sont ainsi offertes aux patients éligibles à la greffe, après l'obtention d'une rémission complète à l'issue du traitement de première ligne (RC1).
Tous les patients ne sont cependant pas éligibles à cette thérapie et certains n'atteignent pas la RC1 avec les traitements conventionnels. C'est dans ce contexte que s'inscrivent d'autres approches thérapeutiques et en particulier les thérapies ciblées.
Traiter la LpDC à l'heure des thérapies ciblées
Les rechutes fréquentes chez les patients ont incité à développer de nouvelles approches thérapeutiques, en particulier par le ciblage de l'antigène caractéristique des blastes leucémiques : le CD123. Cet antigène représente la chaîne α du récepteur à l'interleukine (IL) 3 qui joue un rôle important dans la physiologie des pDC.
La première molécule développée dans le but de cibler le CD123 a été le tagraxofusp (SL-401) en 2014. Il s'agit d'une protéine de fusion entre l'IL-3 humaine et une forme tronquée de la toxine diphtérique. La reconnaissance par le CD123 de son ligand (IL-3) entraîne l'internalisation de la molécule et la libération intracellulaire de la toxine diphtérique qui inhibe la synthèse protéique et entraîne la mort de la cellule cible. La publication initiale s'intéressait à 47 patients qui avaient reçu, ou non, un premier traitement. Parmi les patients n'ayant reçu aucun traitement (29 cas), il a été observé une survie à 2 ans de 54 % avec un taux de rémission complète de 72 % permettant un recours à l'allogreffe dans presque la moitié des cas23. Ceci a permis l'obtention d'une autorisation de mise sur le marché (AMM) aux Etats-Unis en 2018. Les données américaines ont pu être confirmées en « vie réelle » récemment dans un travail publié en 2022 et incluant 22 patients européens chez qui on observe un taux de réponse complète de 67 % avec le tagraxofusp en 1ère ligne de traitement24. Les difficultés rencontrées avec cette thérapie ciblée sont celles de sa toxicité et de la résistance acquise. La principale toxicité rencontrée est le syndrome de fuite capillaire, fréquente (19 %23 à 59 %24 selon les cohortes) et potentiellement grave. Enfin, la résistance potentielle au traitement semble en lien avec une méthylation des gènes impliqués dans l'action de la toxine diphtérique, réversible par l'utilisation d'agents hypométhylants 25.
Le pivekimab sunirine (ou IMGN632) est un anticorps monoclonal couplé à une chimiothérapie de type agent alkylant de l'ADN dont l'efficacité a été montrée chez 23 patients atteints de LpDC en rechute ou réfractaire avec un taux de réponse objectivé de 30 %. Par opposition au tagraxofusp, il présente un profil de sécurité plus favorable avec un schéma d'administration ne nécessitant pas d'hospitalisation complète (une injection toutes les 3 semaines contre 5 jours d'injection toutes les 3 semaines pour le tagraxofusp)26, 27.
Le CD123 peut également être ciblé par l'utilisation des CAR-T (Chimeric Antigen Receptor-T) cells. L'UMR RIGHT de Besançon travaille sur la mise au point d'un CAR-T dirigé contre le CD123 dans la LpDC avec une bonne efficacité dans des modèles précliniques in vivo et in vitro. En effet, la cellule T modifiée est capable d'exercer une cytotoxicité sur les lignées cellulaires disponibles au laboratoire (à savoir CAL-1 et GEN 2-2) mais également sur des échantillons primaires de patient injectés dans des souris immunodéprimées (Patient Derived Xenograft ou PDX)28, 29. Cette technologie est actuellement en transfert vers la clinique et un essai clinique de phase IB devrait être réalisé à moyen terme dans les LpDC.
Autres options thérapeutiques dans la LpDC
Parmi les autres thérapeutiques pouvant avoir une effi cacité dans la LpDC on retrouve le vénétoclax (en lien avec une expression importante de Bcl2 par les cellules tumorales)30, le bortézomib31 ou encore le daratumumab (en lien avec une expression très fréquente du CD38 à la surface des cellules leucémiques)32.

Réseau ROMI et le CHU de Besançon comme laboratoire de biologie médicale de référence
Depuis 2004, le réseau national ROMI piloté par le Pr Francine GARNACHEOTTOU à Besançon a permis la collection d'une importante base d'échantillons biologiques de patients atteints de LpDC et de LAMpDC. Le laboratoire de biologie du CHU de Besançon a obtenu le statut de laboratoire de biologie médicale de référence (LBMR) pour le diagnostic et le suivi biologique de ces hémopathies. Vous pouvez retrouver des informations concernant le réseau sur le site internet du CHU de Besançon* ainsi que les coordonnées permettant d'adresser les cas pour lesquelles une expertise est requise en cas de suspicion d'hémopathie à pDC dans l'un de vos centres ainsi que la feuille de demande d'examen. Une journée sur le thème des hémopathies à pDC sera organisée le vendredi 16 mai à Besançon, à vos agendas !
Messages clés
• La LpDC concerne environ 40 nouveaux patients par an en France.
•Les symptômes cutanés sont les manifestations cliniques les plus fréquentes et constituent dans environ un tiers des cas la seule atteinte de la pathologie.
•Le diagnostic cytologique est complexe et le diagnostic requiert un immunophénotypage par cytométrie en flux à la recherche du profil caractéristique CD123+ CD4+ CD56+. Le diagnostic formel nécessite de confirmer la lignée pDC via l'expression de marqueurs associés comme CD303, CD304, FcER1 et TCL1.
•Les anomalies moléculaires et cytogénétiques associent des anomalies retrouvées dans les pathologies myéloïdes et lymphoïdes sans qu'aucune ne soit spécifique de la LpDC.
• Le pronostic de la maladie est défavorable avec des rechutes chez environ 75 % des patients tout traitement confondu.
• La prise en charge permettant d'obtenir les meilleurs taux de survie chez les patients nécessite l'obtention d'une rémission complète en 1ère ligne afin de proposer une greffe de CSH.
• Les rechutes malgré le recours à la greffe de CSH incitent au développement de nouvelles stratégies thérapeutiques, ciblant en particulier le CD123.
• Une meilleure compréhension de la physiopathologie et des mécanismes de résistance au traitement et de rechutes est nécessaire afin d'améliorer le pronostic de cette maladie.
Références
1. Fitzgeraldbocarsly P, Dai J, Singh S. Plasmacytoid dendritic cells and type I IFN: 50 years of convergent history. Cytokine Growth Factor Rev. févr 2008;19(1):3-19.
2. Ye Y, Gaugler B, Mohty M, Malard F. Plasmacytoid dendritic cell biology and its role in immune-mediated diseases. Clin Transl Immunol [Internet]. janv 2020 [cité 10 mars 2023];9(5). Disponible sur : https://onlinelibrary.wiley.com/doi/10.1002/cti2.1139
3. Garnache-Ottou F, Vidal C, Biichlé S, Renosi F, Poret E, Pagadoy M, et al. How should we diagnose and treat blastic plasmacytoid dendritic cell neoplasm patients? Blood Adv. 23 déc 2019;3(24):4238-51.
4. Laribi K, Baugier De Materre A, Sobh M, Cerroni L, Valentini CG, Aoki T, et al. Blastic plasmacytoid dendritic cell neoplasms: results of an international survey on 398 adult patients. Blood Adv. 13 oct 2020;4(19):4838-48.
5. Cuglievan B, Connors J, He J, Khazal S, Yedururi S, Dai J, et al. Blastic plasmacytoid dendritic cell neoplasm: a comprehensive review in pediatrics, adolescents, and young adults (AYA) and an update of novel therapies. Leukemia. sept 2023;37(9):1767-78.
6. Pagano L, Valentini CG, Pulsoni A, Fisogni S, Carluccio P, Mannelli F, et al. Blastic plasmacytoid dendritic cell neoplasm with leukemic presentation: an Italian multicenter study. Haematologica. 1 févr 2013;98(2):239-46.
7. Pemmaraju N, Wilson NR, Khoury JD, Jain N, Daver N, Pierce S, et al. Central nervous system involvement in blastic plasmacytoid dendritic cell neoplasm. Blood. 14 oct 2021;138(15):1373-7.
8. Herling M, Patel KA, Teitell MA, Konopleva M, Ravandi F, Kobayashi R, et al. High TCL1 expression and intact T-cell receptor signaling define a hyperproliferative subset of T-cell prolymphocytic leukemia. Blood. 1 janv 2008;111(1):328-37.
9. Garnache-Ottou F, Feuillard J, Ferrand C, Biichle S, Trimoreau F, Seilles E, et al. Extended diagnostic criteria for plasmacytoid dendritic cell leukaemia. Br J Haematol. juin 2009;145(5):624-36.
10. Zalmaï L, Viailly PJ, Biichle S, Cheok M, Soret L, Angelot-Delettre F, et al. Plasmacytoid dendritic cells proliferation associated with acute myeloid leukemia: phenotype profile and mutation landscape. Haematologica. 13 oct 2020;106(12):3056-66.
11. Xiao W, Chan A, Waarts MR, Mishra T, Liu Y, Cai SF, et al. Plasmacytoid dendritic cell expansion defines a distinct subset of RUNX1-mutated acute myeloid leukemia. (5).
12. Dress RJ, Dutertre CA, Giladi A, Schlitzer A, Low I, Shadan NB, et al. Plasmacytoid dendritic cells develop from Ly6D+ lymphoid progenitors distinct from the myeloid lineage. Nat Immunol. juill 2019;20(7):852-64.
13. Reizis B. Plasmacytoid Dendritic Cells: Development, Regulation, and Function. Immunity. janv 2019;50(1):37-50.
14. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 30 juill 2009;114(5):937-51.
15. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Myeloid and Histiocytic/Dendritic Neoplasms. Leukemia. juill 2022;36(7):1703-19.
16. Renosi F, Callanan M, Lefebvre C. Genetics and Epigenetics in Neoplasms with Plasmacytoid Dendritic Cells. Cancers. 26 août 2022;14(17):4132.
17. Griffin GK, Booth CAG, Togami K, Chung SS, Ssozi D, Verga JA, et al. Ultraviolet radiation shapes dendritic cell leukaemia transformation in the skin. Nature [Internet]. 7 juin 2023 [cité 8 juin 2023]; Disponible sur : https://www.nature.com/articles/ s41586-023-06156-8
18. Pemmaraju N, Kantarjian HM, Khoury JD, Loghavi S, O'Brien S, Cortes JE, et al. Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN) Commonly Presents in the Setting of Prior or Concomitant Hematologic Malignancies (PCHM): Patient Characteristics and Outcomes in the Rapidly Evolving Modern Targeted Therapy Era. Blood. 13 nov 2019;134(Supplement_1):2723-2723.
19. Batta K, Bossenbroek HM, Pemmaraju N, Wilks DP, Chasty R, Dennis M, et al. Divergent clonal evolution of blastic plasmacytoid dendritic cell neoplasm and chronic myelomonocytic leukemia from a shared TET2-mutated origin. Leukemia. nov 2021;35(11):3299-303.
20. Tang Z, Li Y, Wang W, Yin CC, Tang G, Aung PP, et al. Genomic aberrations involving 12p/ETV6 are highly prevalent in blastic plasmacytoid dendritic cell neoplasms and might represent early clonal events. Leuk Res. oct 2018;73:86-94.
21. Suzuki K, Suzuki Y, Hama A, Muramatsu H, Nakatochi M, Gunji M, et al. Recurrent MYB rearrangement in blastic plasmacytoid dendritic cell neoplasm. Leukemia. juill 2017;31(7):1629-33.
22. Aoki T, Suzuki R, Kuwatsuka Y, Kako S, Fujimoto K, Taguchi J, et al. Long-term survival following autologous and allogeneic stem cell transplantation for blastic plasmacytoid dendritic cell neoplasm. Blood. 4 juin 2015;125(23):3559-62.
23. Pemmaraju N, Lane AA, Sweet KL, Stein AS, Vasu S, Blum W, et al. Tagraxofusp in Blastic Plasmacytoid Dendritic-Cell Neoplasm. N Engl J Med. 25 avr 2019;380(17):1628-37.
24. Deconinck E, Anant M, Manteigas D, Paley C, Riggi M, Herling M, et al. Preliminary Results from an Observational Multicenter Study of Patients with Blastic Plasmacytoid Dendritic Cell Neoplasm Treated with Tagraxofusp in the European Expanded Access Program. Blood. 15 nov 2022;140(Supplement 1):8115-6.
25. Luskin MR, Lane AA. Tagraxofusp for blastic plasmacytoid dendritic cell neoplasm. Haematologica [Internet]. 23 mars 2023 [cité 8 janv 2024]; Disponible sur : https://haematologica.org/article/view/haematol.2022.282171
26. Daver NG, Montesinos P, DeAngelo DJ, Wang ES, Papadantonakis N, Deconinck E, et al. Clinical Profile of IMGN632, a Novel CD123-Targeting Antibody-Drug Conjugate (ADC), in Patients with Relapsed/Refractory (R/R) Acute Myeloid Leukemia (AML) or Blastic Plasmacytoid Dendritic Cell Neoplasm (BPDCN). Blood. 13 nov 2019;134(Supplement_1):734-734.
27. Daver NG, Erba HP, Papadantonakis N, DeAngelo DJ, Wang ES, Konopleva MY, et al. A Phase 1b/2 Study of the CD123-Targeting Antibody-Drug Conjugate IMGN632 As Monotherapy or in Combination with Venetoclax and/or Azacitidine for Patients with CD123-Positive Acute Myeloid Leukemia. Blood. 13 nov 2019;134(Supplement_1):2601-2601.
28. Bôle-Richard E, Fredon M, Biichlé S, Anna F, Certoux JM, Renosi F, et al. CD28/4-1BB CD123 CAR T cells in blastic plasmacytoid dendritic cell neoplasm. Leukemia. déc 2020;34(12):3228-41.
29. Fredon M, Poussard M, Biichlé S, Bonnefoy F, Mantion CF, Seffar E, et al. Impact of scFv on Functionality and Safety of ThirdGeneration CD123 CAR T Cells. Cancer Immunol Res. 1 août 2024;12(8):1090-107.
30. Gangat N, Konopleva M, Patnaik MM, Jabbour E, DiNardo C, Al-Kali A, et al. Venetoclax and hypomethylating agents in older/ unfit patients with blastic plasmacytoid dendritic cell neoplasm. Am J Hematol [Internet]. févr 2022 [cité 27 janv 2024];97(2). Disponible sur : https://onlinelibrary.wiley.com/doi/10.1002/ajh.26417
31. Philippe L, Ceroi A, Bôle-Richard E, Jenvrin A, Biichle S, Perrin S, et al. Bortezomib as a new therapeutic approach for blastic plasmacytoid dendritic cell neoplasm. Haematologica. nov 2017;102(11):1861-8.
32. Pemmaraju N, Kantarjian H, Sweet K, Wang E, Senapati J, Wilson NR, et al. North American Blastic Plasmacytoid Dendritic Cell Neoplasm Consortium: position on standards of care and areas of need. Blood. 9 févr 2023;141(6):567-78.

Valentin POURCHET
Interne en Biologie médicale
au CHU de Besançon
Avec la relecture de
Pr Francine GARNACHE-OTTOU
CHU de Besançon