

Physiopathologie
L'HTP est un phénomène hémodynamique défini par l'élévation de la pression artérielle pulmonaire moyenne (PAPm) au-dessus d'un seuil pathologique, mesurée lors d'un cathétérisme cardiaque droit (CCD). Chez les sujets sains, la PAPm allongée et au repos est de 14 ± 3 mmHg, quel que soit l'âge, le sexe, l'ethnie, etc. Le seuil pathologique est fixé à 20 mmHg soit 2 déviations standard de la norme (2). L'HTP est donc définie par une mesure par CCD d'une PAPm supérieure à 20 mmHg au repos. L'échocardiographie est un bon examen de dépistage mais ne peut en aucun cas être suffisant pour établir le diagnostic d'HTP.
Lors du CCD, sont mesurés également d'autres variables comme la pression auriculaire droite (POD), la PAP occluse (PAPO), le débit cardiaque, et la saturation en oxygène du sang veineux mêlé (SvO2). Les autres variables d'intérêt sont calculées : résistances vasculaires pulmonaires (RVP = [PAPm-PAPO]/ Qc), volume d'éjection systolique, compliance artérielle pulmonaire… La mesure de PAPO permet de distinguer les HTP post-capillaires (associées à une cardiopathie gauche), les plus fréquentes, des HTP pré-capillaires. Ainsi, une PAPO supérieure à 15 mmHg définit une HTP post-capillaire. Enfin, pour caractériser le caractère pré-capillaire d'une HTP, on introduit également la notion de RVP supérieure à 2 UW. Une HTP peut donc être pré-capillaire (PAPm supérieure à 20 mmHg, PAPO inférieure ou égale à 15 mmHg et RVP supérieure à 2 UW), post-capillaire isolée (PAPm supérieure à 20 mmHg, PAPO supérieure ou égale à 15 mmHg et RVP inférieure ou égale à 2 UW), ou bien post- et pré-capillaire combinée (PAPm supérieure à 20 mmHg, PAPO supérieure ou égale à 15 mmHg et RVP supérieure à 2 UW) (Tableau 1). Enfin, sachez qu'il existe également une définition de l'hypertension pulmonaire d'effort, dont le diagnostic est porté par un CCD réalisé pendant un effort à diff érents niveaux de puissance, permettant de construire une pente pression/débit. L'HTP d'effort est défini par une pente PAPm/Qc supérieure à 3 mmHg/L/mn.
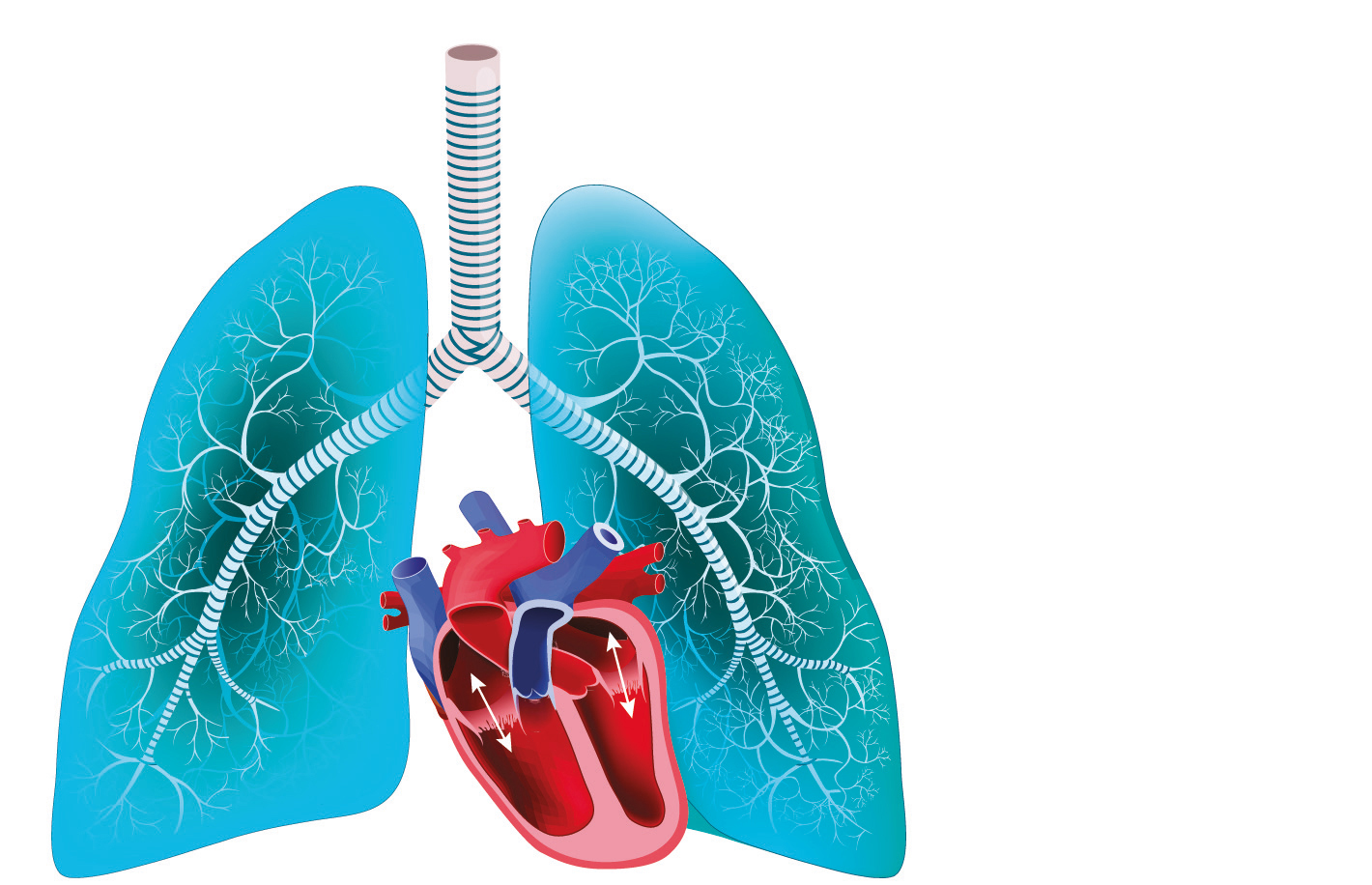
Tableau 1 : Définition hémodynamique des hypertensions pulmonaires et groupes cliniques correspondants dans la classification des recommandations européennes de 2022 (2)
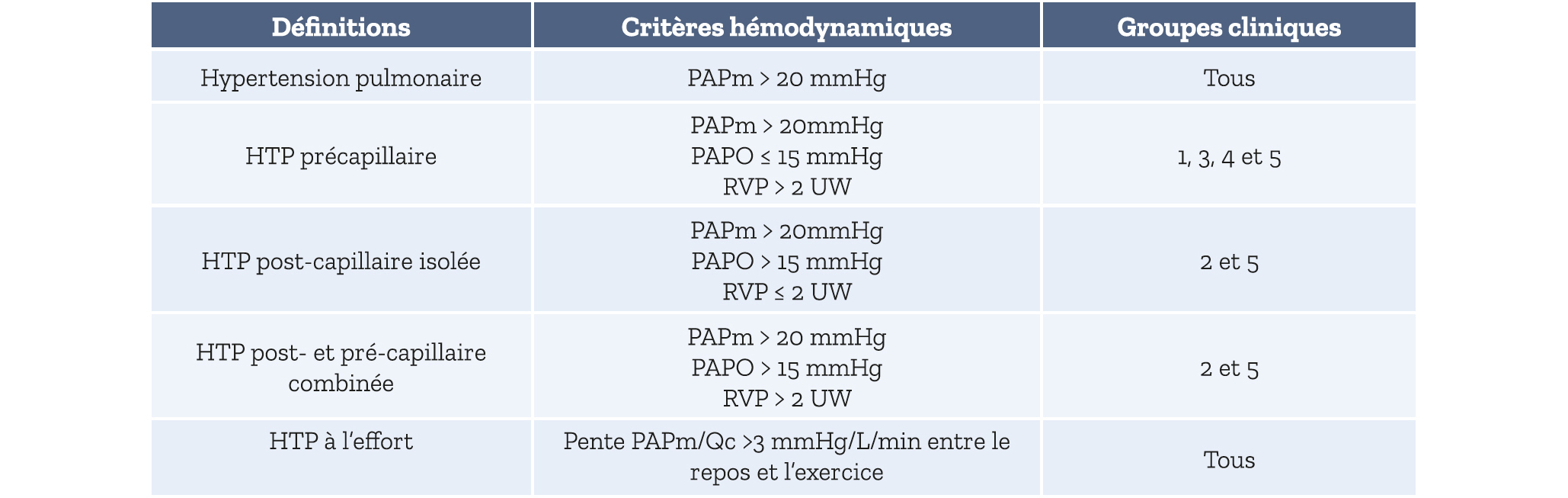
Abréviations : PAPm : pression artérielle pulmonaire moyenne ; PAPO : pression artérielle pulmonaire d'occlusion ; RVP : résistances vasculaires pulmonaires ; UW : unités Wood ; Qc : débit cardiaque.
Classification clinique des hypertensions pulmonaires
De nombreuses pathologies peuvent être associées ou être à l'origine d'une HTP. Elles sont divisées en 5 grands groupes définis par des mécanismes physiopathologiques et donc des stratégies thérapeutiques similaires (Tableau 2). Les HTP post-capillaires isolées ou combinées représentent le groupe 2, alors que les autres groupes abritent les HTP précapillaires (parfois combinées dans le groupe 5).
→ Le groupe 1 correspond à l'hypertension artérielle pulmonaire (HTAP), définit par une hypertension pulmonaire pré-capillaire provoquée par un intense remodelage vasculaire pulmonaire en lien avec des mécanismes complexes et en particulier une dysfonction endothéliale des artères pulmonaires. Il s'agit de maladies rares, qui peuvent être d'origine génétique (une vingtaine de gènes candidats, la mutation la plus fréquente touchant BMPR2, un récepteur impliqué dans la voie du TGF-β), associée à la prise de médicaments ou toxiques (anorexigènes dérivés de la fenfl uramine, dasatinib, méthamphétamine…), ou bien associées à certaines conditions au cours desquelles la prévalence de l'HTAP est plus élevée que dans la population générale : maladies auto-immunes (notamment la sclérodermie systémique), hypertension portale (avec ou sans cirrhose), infection par le VIH, cardiopathies congénitales. Quand on ne trouve aucun facteur favorisant ou maladie associée, on parle d'HTAP idiopathique. On retrouve également dans le groupe 1 les HTAP avec atteinte veinulaire ou capillaire prédominantes (maladie veino-occlusive [MVO] et hémangiomatose capillaire pulmonaire [HCP]) qui sont des HTAP pré-capillaires qui posent cependant des problèmes spécifiques de prise en charge et qui sont de très mauvais pronostic (1).
→ Le groupe 2 correspond aux HTP postcapillaire, c'est-à-dire causées par une pathologie cardiaque gauche. Le phénomène est très fréquent (supérieure à 65 % des cas d'HTP), proportionnel à la sévérité de la cardiopathie (3). La cause la plus fréquente d'HTP postcapillaire est l'insuffisance cardiaque à fraction d'éjection préservée (en anglais, HFpEF pour Heart Failure with preserved Ejection Fraction), de diagnostic parfois difficile à l'échocardiographie. Typiquement, il s'agit de patients âgés avec des comorbidités cardiovasculaires comme l'hypertension, le diabète, l'obésité ou une cardiopathie ischémique.
→ Le groupe 3 regroupe les HTP associées aux maladies respiratoires chroniques (BPCO, emphysème, pneumopathies interstitielles diffuses…) et/ou l'hypoxie chronique. Elles représentent la seconde forme la plus fréquente d'HTP après les HTP post-capillaires. Elles ont souvent une sévérité modérée, corrélée à la gravité de la maladie causale, mais elles peuvent être parfois très sévères avec un profil "vasculaire" sans lien avec la sévérité de l'atteinte respiratoire.
→ Le groupe 4 est défini par une obstruction des artères pulmonaires, généralement secondaire à une maladie thrombo-embolique veineuse chronique, ou plus rarement par des tumeurs vasculaires ou des artérites.
→ Enfin, on place dans le groupe 5 les pathologies responsables d'HTP de mécanismes incertains et/ou multifactoriels. On peut y citer par exemple la sarcoïdose, la drépanocytose ou l'insuffisance rénale chronique sévère.
La composition des groupes évolue régulièrement, au gré des avancées dans la compréhension des mécanismes physiopathologiques et des connaissances épidémiologiques. Les modifications les plus récentes sont la refonte du groupe 3 pour bien diff érencier des entités cliniques (BPCO, PID, etc.) et non plus des troubles ventilatoires, ou bien la suppression du syndrome d'apnées du sommeil qui n'est pas en soi une cause d'HTP en l'absence d'hypoventilation alvéolaire chronique, ou encore la distinction au sein des HTAP idiopathiques d'un sous-groupe de très bon pronostic, avec une vasoréactivité en aigu et répondant favorablement au long-cours aux inhibiteurs calciques, et l'ajout au groupe 1 des HTAP avec atteinte veinulaires et/ou capillaires (MVO et HCP).
Tableau 2 : Dernière version de la classification clinique des hypertension pulmonaires, d'après les recommandations d'experts du 7ème symposium mondial sur l'hypertension pulmonaire (WSPH 2024) (4)

Abréviations : HTAP : hypertension artérielle pulmonaire ; HTP : hypertension pulmonaire ; MVO : maladie veino-occlusive ; HCP : hémangiomatose capillaire pulmonaire ; BPCO : bronchopneumopathie chronique obstructive ; # : certains patients avec une HTAP héritable ou une HTAP associée à des médicaments et toxiques peuvent être répondeurs au long-cours aux inhibiteurs calciques ; ¶ : cardiopathie hypertrophique, amylose, maladie de Fabry, maladie de Chagas ; + : maladies parenchymateuses non incluses dans le groupe 5 ; § : les autres causes d'obstruction artérielle pulmonaire incluent les sarcomes (de grade élevé ou intermédiaire, ou les angiosarcomes), les autres tumeurs malignes (par exemple, carcinome rénal ou utérin, tumeurs germinales testiculaires), les tumeurs bénignes (par exemple le léiomyome utérin), les artérites sans connectivite, les sténoses congénitales des artères pulmonaires, et les hydatidoses ; * : anémies hémolytiques chroniques constitutionnelles ou acquises, syndromes myéloprolifératifs chroniques ; ## : glycogénoses, maladie de Gaucher.

Prise en charge d'un patient avec suspicion d'hypertension pulmonaire
Maintenant que vous savez définir les diff érents types d'hypertension pulmonaire, comment les reconnaître et les prendre en charge en pratique ? Nous vous proposons de détailler l'algorithme en 3 grandes étapes.
Étape 1 : la suspicion clinique
Devant des symptômes évocateurs (dyspnée d'effort inexpliquée le plus souvent), dès l'interrogatoire, certains indices simples peuvent vous faire évoquer la possibilité d'une HTP : un contexte de connectivite ou d'hypertension portale, un antécédent familial connu d'HTP, ou des épisodes d'embolie pulmonaire / phlébites à répétition par exemple.
Les signes cliniques en lien avec l'HTP sont aspécifiques : le motif de consultation est généralement une dyspnée d'effort chronique, souvent depuis plusieurs mois, d'aggravation progressive. Quelle que soit l'étiologie, on peut retrouver des signes d'insuffisance cardiaque droite, des douleurs thoraciques atypiques ou des histoires de malaises voire de syncopes à l'effort (signe de gravité). L'auscultation cardiaque peut révéler un éclat du B2 au foyer pulmonaire, des arythmies responsables de palpitations, et surtout un souffl e systolique d'insuffisance tricuspide. Le reste de votre examen clinique pourra vous orienter vers la maladie causale (anomalie de l'auscultation pulmonaire, signes de connectivite, hypertension portale, etc.).
Étape 2 : la détection
Il est généralement indiqué chez tout patient avec une dyspnée d'effort chronique non expliquée par une pathologie respiratoire (BPCO, asthme…) ou une anémie chronique, de réaliser une échographie cardiaque transthoracique. C'est cet examen qui, en plus de rechercher une cardiopathie gauche, permet de dépister une hypertension pulmonaire et de donner 3 niveaux de probabilité en fonction de la vitesse maximale du fl ux de régurgitation tricuspide (VmaxIT). La VmaxIT permet d'estimer (et non pas de mesurer) la pression artérielle pulmonaire systolique (PAPS) qui est calculée à partir de la VmaxIT (PAPS = 4 x VmaxIT²). La probabilité d'HTP est faible en dessous de 2,8 m/s, forte au-dessus de 3,4 m/s et intermédiaire entre les deux.
Il est aussi recommandé d'évaluer les signes indirects d'HTP tels qu'une dilatation ou hypertrophie du ventricule droit, un défaut de contractilité, un septum paradoxal… Il ne faut donc jamais se contenter d'une PAPS estimée comme seule information disponible pour suspecter le diagnostic d'HTP.
On peut retrouver des signes d'HTP dans les autres examens classiquement réalisés dans l'exploration des dyspnées chroniques :
→ L'électrocardiogramme (ECG) peut montrer une déviation axiale droite, un bloc de branche droit et/ou une hypertrophie ventriculaire droite.
→ La biologie recherche une augmentation des facteurs natriurétiques auriculaires comme le BNP (Brain natriuretic peptide) et/ou le NT-proBNP (N-terminal pro-brain natriuretic peptide).
→ Les épreuves fonctionnelles respiratoires (EFR), incluant des gaz du sang, peuvent révéler une pathologie respiratoire dans le cadre d'une HTP du groupe 3. Dans l'HTAP, seule la DLCO est en général modérément diminuée, avec des patients peu hypoxémiques et souvent hypocapniques. En l'absence de pathologie respiratoire (emphysème, PID), une altération profonde de la DLCO doit faire évoquer une MVO.
→ La radiographie thoracique peut montrer une cardiomégalie et un bombement des cavités droites, une hypertrophie hilaire par dilatation des artères pulmonaires, ou une maladie parenchymateuse causale.
→ Au scanner thoracique, on peut voir une dilatation de l'artère pulmonaire, des lésions thrombo-emboliques chroniques (si injection) ou des signes d'une maladie pulmonaire sous-jacente.
Dans tous les cas, une suspicion d'HTP sévère doit faire référer votre patient à un centre de référence ou de compétences du réseau maladies rares ‘PulmoTension' qui regroupe l'ensemble des centres français dédiés à la prise en charge des HTP.
Il s'agit d'une urgence en cas de dyspnée majeure (classe fonctionnelle NYHA 4), de symptômes d'évolution rapide, en cas de syncope ou d'arythmie, ou d'instabilité hémodynamique.
Étape 3 : la confirmation
La confirmation du diagnostic et le bilan étiologique sont réalisés dans un centre de référence ou de compétences et comportent :
→ Un cathétérisme cardiaque droit, seul examen permettant de confirmer le diagnostic d'HTP et d'en apprécier le mécanisme pré- ou post-capillaire. Il permet également de tester la vasoréactivité en cas d'HTAP supposée idiopathique, et d'évaluer certains critères de sévérité, ce qui permettra de guider la conduite thérapeutique.
→ Une scintigraphie pulmonaire de ventilation/perfusion, seul examen permettant d'écarter avec certitude une origine thrombo-embolique chronique.
→ Un scanner thoracique injecté au temps artériel pulmonaire, qui cherchera des arguments pour une maladie parenchymateuses ou des lésions thrombo-emboliques.
→ Une échocardiographie à la recherche d'éléments en faveur d'une cardiopathie congénitale ou d'une cardiopathie gauche.
→ Un bilan biologique comportant un bilan auto-immun, et des sérologies des hépatites B et C et du VIH.
→ Une échographie abdominale avec recherche d'hypertension portale.
→ D'autres examens indiqués en fonction du contexte clinique.
→ La biopsie pulmonaire est toujours contre-indiquée dans l'HTP sévère.
Dans le même temps, on évaluera la sévérité de votre patient avec :
→ L'appréciation de la classe fonctionnelle de la NYHA.
→ La recherche de signe de gravité clinique vus plus haut : aggravation rapide, syncopes, instabilité hémodynamique…
→ Une évaluation de sa capacité à l'effort par un test de marche de 6 minutes, plus rarement par épreuves fonctionnelles à l'exercice.
→ La mesure de biomarqueurs pronostiques : BNP ou NT-proBNP, créatinine, uricémie, bilan hépatique…
→ Et bien sûr les variables échocardiographiques (présence d'un épanchement péricardique, évaluation de la fonction ventriculaire droite, dilatation de l'oreillette droite…) et hémodynamiques (débit et index cardiaques, volume d'éjection systolique, SvO2).
Toute suspicion d'HTP sévère, quelle que soit la cause suspectée, ou suspicion d'HTAP (HTP du groupe 1) ou d'HTP thrombo-embolique chronique (groupe 4) justifie d'un contact rapide avec un centre de référence ou de compétences maladies rares du réseau PulmoTension.
Nous espérons que cet article vous aidera dans votre pratique pour ne manquer aucune hypertension pulmonaire et avoir les bons réfl exes au quotidien. N'hésitez pas à vous renseigner sur le centre de référence ou de compétences le plus proche de votre lieu d'exercice !
Références
1. Hoeper MM, Humbert M, Souza R, Idrees M, Kawut SM, Sliwa-Hahnle K, et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016; 4: 306-22.
2. Humbert M, Kovacs G, Hoeper MM, Badagliacca R, Berger RMF, Brida M, et al. 2022 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Respir J. 2023; 61: 2200879
3. Maron BA, Bortman G, De Marco T, Huston JH, Lang IM, Rosenkranz SH, et al. Pulmonary hypertension associated with left heart disease. Eur Respir J. 2024; 64: 2401344.
4. Kovacs G, Bartolome S, Denton CP, Gatzoulis MA, Gu S, Khanna D, et al. Definition, classification and diagnosis of pulmonary hypertension. Eur Respir J. 2024; 64: 2401324.
Dr Antoine BEAUVAIS, Pr Olivier SITBON
1Université Paris-Saclay, Faculté de Médecine, Le Kremlin-Bicêtre.
2APHP, GHU Paris-Saclay, Hôpital Bicêtre, Centre coordonnateur des centres de référence de l'hypertension pulmonaire (PulmoTension), service de pneumologie et de soins intensifs respiratoires. Le Kremlin-Bicêtre.
3INSERM, UMR_S999 (HPPIT : Hypertension Pulmonaire : Physiopathologie et Innovation Thérapeutique), Le Kremlin-Bicêtre.