

Bénédicte Piron1, Enzo Frankowski1, Camille Debord2, Caroline Mayeur-Rousse1
1 Service d'hématologie clinique, Centre Hospitalier Universitaire de Nantes, Nantes, France.
2 Service d'hématologie biologie, Centre Hospitalier Universitaire de Nantes, Nantes, France.
Merci aux Drs Camille Debord et Caroline Mayeur-Rousse (hémato-biologistes au CHU de Nantes), pour leur relecture attentive.
Nous rapportons le cas d'un patient âgé de 74 ans, vu à plusieurs reprises en hôpital de jour d'hématologie de l'hôpital de Nantes, pour avis concernant une pancytopénie.
Il s'agit d'un patient dont les antécédents médicaux sont marqués essentiellement par un carcinome à cellules claires du rein gauche traité par radiofréquence (six ans auparavant), une cardiopathie ischémique et un AVC ischémique.
Chez ce patient, par l'intermédiaire d'un bilan sanguin systématique, il a été démasqué une pancytopénie avec une anémie normocytaire arégénérative (hémoglobine à 11,5 g/dL), une thrombopénie (plaquettes 97 G/L) et une neutropénie avec des leucocytes à 8,94G/L dont 0,46 G/L de neutrophiles et 0,68 G/L de grands lymphocytes à grains (LGL). Le bilan sanguin un an auparavant était normal avec 13,6 g/dl d'hémoglobine, 205 G/L plaquettes et 6,05 G/L de leucocytes avec une formule normalement équilibrée.

Figure 1. Immunophénotypage des lymphocytes T : population T monoclonale représentant 30% des lymphocytes T totaux
(en rose) : sCD3+ CD5+faible CD8+ CD56- CD16 + CD57+ TCRαβ+ TRBC2+.
Sur le myélogramme réalisé, il a été retrouvé une moelle de richesse subnormale, sans excès de blastes, sans dystrophie, sans cellules anormales mais il a été noté la présence d'une hyperlymphocytose médullaire (24 %) d'allure polymorphe. Un immunophénotypage des lymphocytes a été réalisé ne révélant pas de population aberrante lymphoïde B, mais par contre, une population lymphoïde T anormale CD3+ CD5dim CD7- CD4- CD8+ CD16- CD56- TCRαβ+ exprimant un marqueur de cytotoxicité CD57+ avec restriction d'expression du TRBC2 et représentant environ 30 % des lymphocytes totaux (figure 1). Le caryotype était normal mais la recherche de clonalité T s'est avérée positive sans mutations STAT3/STAT5B identifiées. Ce profil moléculaire, vu les éléments cytologiques (neutropénie et présence de lymphocytes à grain supérieure à 0.5 G/L sur le frottis) et phénotypiques, était donc en faveur d'une leucémie à LGL-T. Le reste du bilan clinico-biologique était sans appel. La neutropénie est restée persistante et sévère, le patient a donc été traité par cyclophosphamide à raison de 100 mg/j, deux mois après le diagnostic.
Un mois après le début du traitement, il est hospitalisé en urgence sur un tableau de syndrome hémorragique cutané associé à un méléna. Sur le plan biologique, on retrouve la persistance d'une pancytopénie avec une leucopénie à 0,71 GL, une hémoglobine à 7,1 g/dl, des plaquettes à 33 G/L associée à une coagulation intravasculaire disséminée avec un TP à 54 %, un fibrinogène à 0,8 g/L et des D-dimères à 23 247 ng/mL.
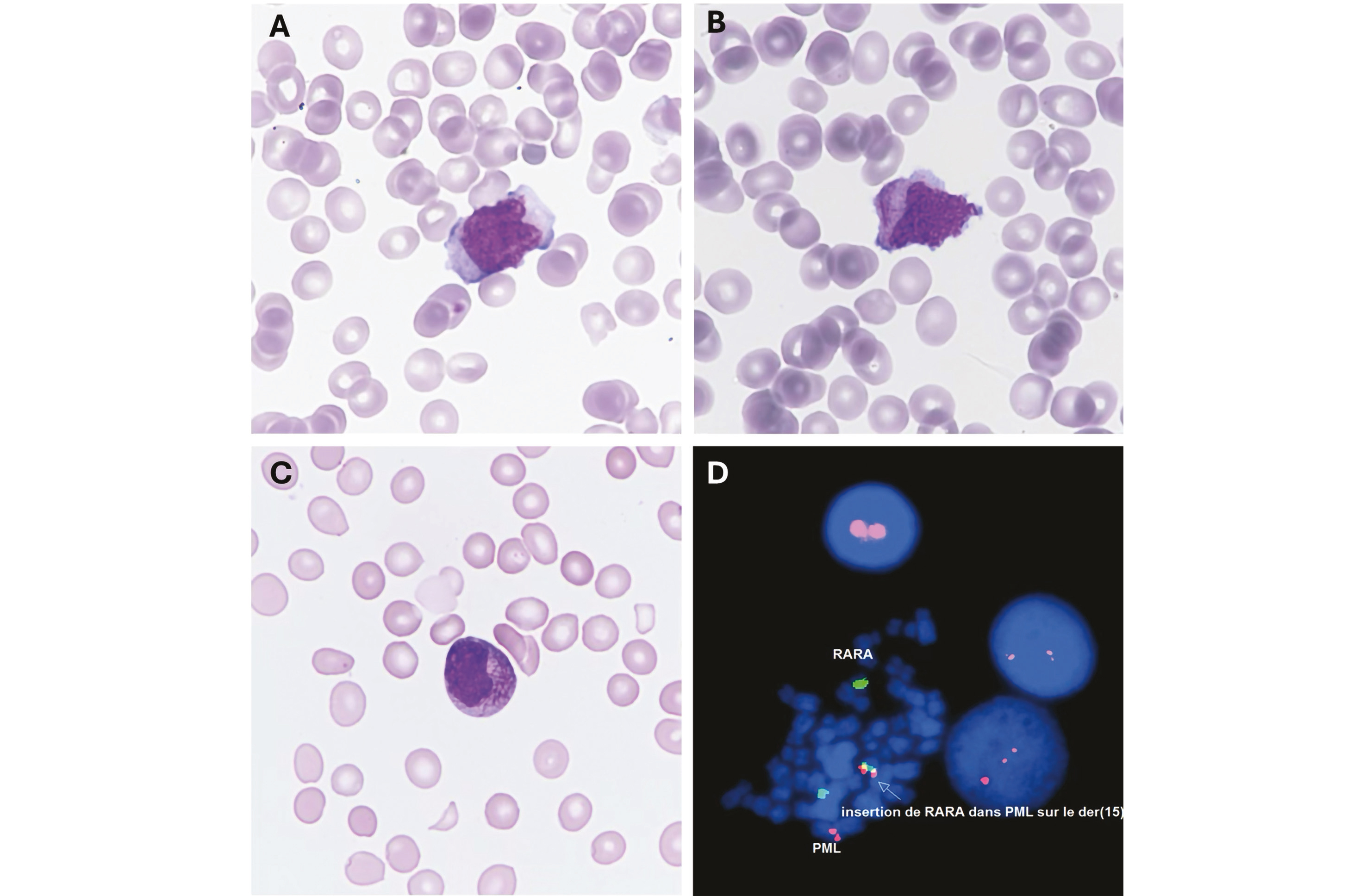
Figure 2. Leucémie aiguë promyélocytaire. A : Frottis sanguin retrouvant un blaste en aile de papillon, grossissement x50 au May-Grünwald-Giemsa (MGG). B : Frottis sanguin retrouvant un blaste avec un corps d'Auer, grossissement x50 au MGG. C : Frottis médullaire retrouvant un blaste avec des corps d'Auer en fagots, grossissement x50 au MGG. D : Réarrangement PML::RARA en fluorescence in situ hybridation (FISH).
Un myélogramme est réalisé retrouvant 87 % de blastes présentant des fagots d'Auer en faveur d'une leucémie aiguë promyélocytaire (LAP) ou LAM 3 de la classification FAB (figure 2 A-C). Ce diagnostic a été confirmé par la présence d'un réarrangement PML::RARA, par translocation t(15;17)(q24;q21), sur 80 % des noyaux observés (figure 2 D). Le patient a été traité selon le protocole de Burnett et al. [1] (all-trans retinoic acid (ATRA) et arsenictrioxide) et est actuellement en réponse complète, en cours de traitement de consolidation.
L'analyse rétrospective par FISH du prélèvement médullaire réalisé trois mois auparavant, a retrouvé un réarrangement PML::RARA dans une faible population cellulaire, sur 5 % des noyaux observés, témoignant de la présence a minima de la LAP à la pancytopénie présentée initialement.
Les LGL-T étaient donc bien clonaux comme confirmé en cytométrie en flux et en biologie moléculaire mais probablement « réactionnels » (pas de mutations retrouvées en next generation sequencing (NGS)).
Ces LGL-T clonaux dirigés contre le compartiment myéloïde pathologique ont probablement permis initialement de contrôler le clone promyélocytaire. Ces derniers éliminés par la prise de cyclophosphamide ont permis de faire émerger la population blastique promyélocytaire sous-jacente.
Discussion
Les associations entre leucémie LGL-T et autres hémopathies malignes ont été décrites dans la littérature. Elles concernent tant les hémopathies lymphoïdes (notamment gammapathie monoclonale de signification indéterminée, leucémie lymphoïde chronique, myélome multiple) [2] que myéloïdes (syndrome myélodysplasique, néoplasies myéloprolifératives) [3, 4]. Cependant, l'association spécifique LAP/LGL-T n'a été décrite qu'une seule fois [5]. Dans cet autre cas rapporté [5], la population LGL-T avait été découverte a posteriori, et contrairement à notre cas, celle-ci avait progressé en parallèle de la rémission du clone blastique. Ainsi, s'il est décrit une prolifération des clones LGL lors du contrôle spécifique des hémopathies concomitantes, l'immunosuppression conduisant à une déplétion des LGL semble favoriser l'échappement leucémique [6].
En effet, cette coexistence de clones LGL et hémopathies myéloïdes peut s'expliquer par 3 types de scénarios différents [7] :
- Coexistence « simultanée » : phénomènes inflammatoires et modifications génétiques acquises parallèlement au vieillissement, font apparaître concomitamment une population clonale de LGL et une population myéloïde clonale.
- Évènement secondaire : la population clonale LGL a un rôle d'immunosurveillance et contrôle la présence de cellules souches hématopoïétiques normales (responsable des cytopénies) et des cellules souches aberrantes (responsables en cas d'immunoévasion, de l'apparition de l'hémopathie myéloïde).
- Évènement primaire : la population monoclonale LGL exerce un rôle cytotoxique inflmmatoire pathogénique sur les cellules souches hématopoïétiques, causant des dommages dans le compartiment myéloïde et favorisant le développement d'une hémopathie myéloïde.
Au contraire de la LAP, la leucémie à LGL reste d'évolution indolente. Ainsi, si la leucémie à LGL-T, est caractérisée par la présence fréquente d'une anémie, moins fréquemment d'une thrombopénie et plus communément d'une neutropénie (inférieure à 1.5 G/L chez 80 % des patients et parfois sévère i.e. inférieure à 0.5 G/L chez 20-25% des patients), celle-ci ne doit pas faire méconnaître une autre hémopathie.
Ainsi, ce cas clinique rappelle la nécessaire vigilance quant aux deux critères obligatoires diagnostiques de LGL-T, rappelés ici [8] :
- La présence au frottis sanguin d'une prolifération clonale de LGL supérieure à 0.5 G/L, pendant au moins 6 mois, sur la base d'une numération globulaire complète et/ou d'un profil phénotypique compatible. L'immunophénotypage retrouve ainsi une prolifération T cytotoxique effectrice CD8+, CD3+, TCRαβ+, CD57+, CD16+(80%), CD56 -/+ et CD5+faible. Dans 20 % des cas, cette population T est TCRɣδ+, CD4-, CD8-/+ ou TCRαβ +, CD4+, CD8- et dans de rares cas : TCRαβ+, CD4+, CD8+ ou CD4-, CD8- .
- Une prolifération monoclonale en cytométrie en flux (CMF) ou en biologie moléculaire. La recherche de clonalité, indispensable, implique en CMF l'étude du répertoire TCRβ ou de la restriction d'expression de la partie constante de la chaîne Béta du TCR (TRBC1/TRBC2). En biologie moléculaire, il convient de faire la preuve d'une clonalité T. La présence de mutations récurrentes comme STAT3/STAT5B viennent conforter le diagnostic de leucémie à LGL-T car ces critères de « prolifération clonale » en CMF ou en biologie moléculaire peuvent conduire à l'identification de clones lymphoïdes T de signification indéterminée (TCUS) [9].
Références
1. Burnett AK, Russell NH, Hills RK, Bowen D, Kell J, Knapper S, et al. Arsenic trioxide and all-trans retinoic acid treatment for
acute promyelocytic leukaemia in all risk groups (AML17): results of a randomised, controlled, phase 3 trial. Lancet Oncol. oct
2015;16(13):1295-305.
2. Cheng J, Talamo G, Malysz J, Ochmann M, Lamy T, Loughran TP. Report of 6 Cases of Large Granular Lymphocytic Leukemia
and Plasma Cell Dyscrasia. Clin Lymphoma Myeloma Leuk. Oct 2014;14(5):e169-72.
3. Lamy T, Loughran TP. How I treat LGL leukemia. Blood. 10 mars 2011;117(10):2764-74.
4. Savaşan S, Al‐Qanber B, Buck S, Wakeling E, Gadgeel M. Clonal T‐cell large granular lymphocyte proliferations in childhood
and young adult immune dysregulation conditions. Pediatr Blood Cancer. mai 2020;67(5):e28231.
5. Reda G, Fattizzo B, Cassin R, Flospergher E, Orofino N, Gianelli U, et al. Multifactorial neutropenia in a patient with acute
promyelocytic leukemia and associated large granular lymphocyte expansion: A case report. Oncol Lett. mars 2017;13(3):1307-10.
6. Fattizzo B, Bellani V, Pasquale R, Giannotta JA, Barcellini W. Large Granular Lymphocyte Expansion in Myeloid Diseases and
Bone Marrow Failure Syndromes: Whoever Seeks Finds. Front Oncol. 1 oct 2021;11:748610.
7. Calabretto G, Attardi E, Gurnari C, Semenzato G, Voso MT, Zambello R. LGL Clonal Expansion and Unexplained Cytopenia: Two
Clues Don't Make an Evidence. Cancers. 25 oct 2022;14(21):5236.
8. Marchand T, Lamy T, Loughran TP. A modern view of LGL leukemia. Blood. 31 oct 2024;144(18):1910-23.
9. Shi M, Olteanu H, Jevremovic D, He R, Viswanatha D, Corley H, et al. T-cell clones of uncertain significance are highly prevalent and show close resemblance to T-cell large granular lymphocytic leukemia. Implications for laboratory diagnostics. Mod Pathol. oct 2020;33(10):2046-57.

Benedicte PIRON
Interne en Hématologie
CHU de Nantes