


Mirtazapine as Appetite Stimulant in Patients with Non–Small Cell Lung Cancer and Anorexia, A Randomized Clinical Trial
O. Arrieta et al, JAMA Oncology 2024.
Actuellement, il n'existe pas de traitement validé pour améliorer l'anorexie liée au cancer. La dénutrition qui en résulte à un impact majeur sur la survie des patients. Dans l'étude présentée ici, la Mirtazapine a été évalué comme traitement pouvant permettre de diminuer l'anorexie des patients pris en charge pour un cancer. Il s'agit d'évaluer l'effet de la Mirtazapine sur l'appétit et la consommation d'énergie chez les patients atteints de cancer du poumon non à petites cellules (NSCLC) avancé. Il s'agit d'un médicament de la famille des antidépresseur, antagoniste au niveau des récepteurs présynaptiques α2 centraux, des récepteurs postsynaptiques 5HT2/5HT3 et des récepteurs H1.
Cette étude clinique randomisée, en double aveugle, contrôlée par placebo, a été réalisée chez des adultes atteints de NSCLC avancé d'août 2018 à mai 2022, avec un suivi de 8 semaines. Les patients étaient randomisés avec un ratio de 1 :1 pour recevoir soit de la Mirtazapine à 15 mg par jour, ou un placebo, pendant 2 semaines, suivies d'une escalade de dose à 30 mg par jour jusqu'à la semaine 8, ou un placebo. Les deux groupes ont reçu une évaluation nutritionnelle et des conseils diététiques. L'appétit a été évalué par l'échelle d'anorexie-cachexie et l'apport énergétique. Les paramètres diététiques ont été évalués au départ, à 4 et 8 semaines, avec un rappel diététique de 24 heures et une quantification énergétique basée sur le système mexicain d'équivalents nutritionnels.
Au total, 86 patients ont rempli les critères d'inclusion et ont été randomisés dans le groupe placebo (n = 43) ou le groupe Mirtazapine (n = 43). L'âge moyen était de 63,5 ans, avec 57,7 % de femmes. Les caractéristiques de base étaient similaires entre les groupes. Il n'y avait aucune différence dans les scores d'appétit chez les patients ayant reçu de la Mirtazapine ou un placebo après 4 et 8 semaines. Après 4 semaines, la Mirtazapine a augmenté de manière significative l'apport énergétique (379,3 kcal ; IC à 95 %, 138,2-576,1 ; p inférieure à 0,001), y compris les protéines (22,5 g ; IC à 95 %, 11,5-33,4 ; p = 0,001), les glucides (43,4 g ; IC à 95 %, 13,1-73,8 ; p = 0,006) et les lipides (13,2 g ; IC à 95 %, 6,0-20,4 ; p = 0,006). L'apport en graisses était significativement plus élevé chez les patients du groupe Mirtazapine (14,5 g vs 0,7 g ; p = 0,02) après 8 semaines. Le groupe Mirtazapine a significativement réduit la proportion de patients présentant une sarcopénie (82,8 % vs 57,1 %, p = 0,03) à 8 semaines. Les patients sous Mirtazapine ont rapporté une bonne tolérance du traitement, mais ont signalé une perception plus élevée des cauchemars à 2 semaines, effet indésirable connu de la Mirtazapine.
Plusieurs limites à souligner dans cet article. Le suivi rapproché et les conseils prodigués lors de l'accompagnement des patients inclus dans l'étude a probablement créé un biais par rapport à un suivi standard des traitements pris en charge pour un cancer. Cela peut expliquer en partie des différences non significatives entre les groupes Mirtazapine et placebo. En effet, soulignons qu'il existe une amélioration au sein du groupe Mirtazapine avec le temps sur les points suscités (apport énergétique en protéine, glucide, lipides), mais comparativement au groupe placebo, la différence n'est pas significative.
Au total dans cet essai, il n'y a aucune différence dans les scores d'appétit chez l'ensemble des patients ayant reçu soit la Mirtazapine ou un placebo, mais le groupe Mirtazapine a présenté une augmentation significative de l'apport énergétique avec le temps sur les 4 et 8 semaines de suivi. L'ajout de la Mirtazapine dans le traitement des patients atteints de NSCLC avancé et d'anorexie pourrait aider ces patients à atteindre leurs besoins énergétiques, cependant les données de cet article ne sont pas suffisamment convaincantes pour que cela devienne un standard en pratique courante.
Long-term patient-reported outcomes from monarchE: Abemaciclib plus endocrine therapy as adjuvant therapy for HR+, HER2-, node-positive, high-risk, early breast cancer.
S. M. Tolaney et al, ECJ 2024
Récemment de nombreuses études étudient quel serait le meilleur traitement adjuvant dans le cancer du sein avec récepteurs hormonaux positifs (RH+). Dans l'étude MonarchE, l'Abémaciclib a démontré un bénéfice durable en termes de survie sans maladie invasive et un profil de sécurité tolérable, avec un suivi médian de 42 mois. Notons que nous n'avons toujours pas les données en survie globale. En l'absence de symptômes liés à la maladie, les thérapies adjuvantes ne doivent pas ou peu altérer la qualité de vie. En effet, de nombreuses patientes sont d'ores et déjà guéries par la chirurgie. L'étude que nous présentons s'est intéressée à évaluer la qualité de vie de ces patientes traitées pendant 2 ans en adjuvant dans l'étude MonarchE.
Les patientes ont rapporté les données de qualité de vie, au démarrage du traitement adjuvant puis à 3, 6, 12, 18 et 24 mois pendant le traitement, et à 1, 6 et 12 mois après l'arrêt du traitement. À noter qu'aucun test statistique n'a été effectué. Au départ, les taux de complétion des instruments PRO étaient supérieure à 96 %. Dans le bras Abémaciclib, des différences significatives pour la diarrhée ont été observées à 3 et 6 mois (augmentations moyennes de 1,19 et 1,03 points sur une échelle de 5 points, respectivement). Cependant, on observe un retour à l'état de base à l'arrêt du traitement, l'effet indésirable est donc réversible. La diminution de la gêne causée par la diarrhée au-delà de 6 mois est probablement dû à l'apprentissage et une meilleure gestion de cet effet. Cet effet indésirable est particulièrement bien connu de la molécule. Il s'agit cependant d'un effet gérable la plupart du temps par des soins de support adapté et traitement anti-diarrhéique. Notons également que les patientes rapportent moins de flush, effet indésirable connu et invalidant de l'hormonothérapie, dans le bras Abémaciclib. Globalement, pendant le traitement, la plupart des patientes dans les deux bras (69 à 78 %) ont déclaré être gênés "un peu" ou "pas du tout" par les effets indésirables. Dans l'ensemble, la fatigue était similaire entre les bras avec ou sans Abémaciclib. Pendant le suivi post-traitement (au-delà de 2 ans), les données de qualité de vie dans les deux bras étaient similaires au départ, c'est-à-dire une réversibilité complète des effets de l'Abémaciclib sur la qualité de vie.
En conclusion, les résultats de qualité de vie confirment un profil de toxicité tolérable, rassurant et réversible pour l'Abémaciclib. La qualité de vie a été préservée avec l'ajout de l'Abémaciclib adjuvant à la thérapie endocrinienne (bloqueurs des récepteurs hormonaux), ce qui conforte l'intérêt de son utilisation chez les patientes atteintes de cancer du sein précoce à haut risque RH+ et HER2-. Précisons à nouveau que les données en survie globale ne sont pas encore matures, critère le plus robuste pour la pertinence d'un traitement adjuvant.
A Cell-free DNA Blood-Based Test for Colorectal Cancer Screening
D.C. Chung et al, NEJM 2024
En introduction, rappelons que le cancer colorectal est le troisième cancer le plus diagnostiqué chez les adultes aux États-Unis. La détection précoce pourrait prévenir plus de 90 % des décès liés au cancer colorectal, pourtant plus d'un tiers de la population éligible au dépistage n'est pas à jour malgré les multiples tests disponibles. Cette proportion est encore plus importante en France ! Un test sanguin à base d'ADN circulant (cfDNA) pourrait permettre d'améliorer l'adhérence au dépistage par la population, de détecter le cancer colorectal plus tôt et de réduire la mortalité liée au cancer colorectal par une prise en charge précoce.
Cette étude épidémiologique évalue les caractéristiques de performance d'un test sanguin à base de cfDNA dans une population éligible au dépistage du cancer colorectal, c'est-à-dire les patients à risque moyen du fait de leur âge (supérieure à 45 ans dans cette étude). Les critères de jugement primaires étaient au nombre de deux, avec premièrement la sensibilité pour le cancer colorectal, et deuxièmement la spécificité pour les lésions néoplasiques avancées (cancer colorectal ou lésions précancéreuses avancées) par rapport à la coloscopie de dépistage. Notons ici que les patients avaient un dépistage invasif par coloscopie et non pas par un test immunologique comme pratiqué dans le dépistage organisé en France. Le critère de jugement secondaire était la sensibilité pour détecter les lésions précancéreuses avancées.
La cohorte de validation clinique comprenait 10258 personnes, dont 7861 remplissaient les critères d'éligibilité et étaient évaluables. Au total, 83,1 % des participants avec un cancer colorectal détecté par coloscopie avaient un test cfDNA positif et 16,9 % avaient un test négatif, ce qui indique une sensibilité du test cfDNA pour la détection du cancer colorectal de 83,1 % (avec un intervalle de confiance à 95 % [IC], 72,2 à 90,3). La sensibilité pour le stade I, II ou III du cancer colorectal était de 87,5 % (IC à 95 %, 75,3 à 94,1), et la sensibilité pour les lésions précancéreuses avancées était de 13,2 % (IC à 95 %, 11,3 à 15,3).
Au total, 89,6 % des participants sans néoplasie colorectale avancée (cancer colorectal ou lésions précancéreuses avancées) identifiée lors de la coloscopie avaient un test sanguin cfDNA négatif, tandis que 10,4 % avaient un test sanguin cfDNA positif, ce qui indique une spécifi cité pour toute néoplasie avancée de 89,6 % (IC à 95 %, 88,8 à 90,3). La spécificité pour une coloscopie négative (pas de cancer colorectal, de lésions précancéreuses avancées ou de lésions précancéreuses non avancées) était de 89,9 % (IC à 95 %, 89,0 à 90,7).
En conclusion, dans une population de dépistage à risque moyen, ce test sanguin à base de cfDNA a environ une sensibilité de 83 % pour le cancer colorectal, une spécificité de 90 % pour les néoplasies avancés et une sensibilité de 13 % pour les lésions précancéreuses avancées. Cette étude ne prenait pas en compte le coût du test qu'il serait intéressant d'évaluer. Néanmoins, un test limité à une prise de sang semblerait tout à fait acceptable et pourrait probablement permettre d'élargir le nombre de personnes dépistées.
Gemcitabine and Paclitaxel Versus Gemcitabine Alone After 5-Fluorouracil, Oxaliplatin, and Irinotecan in Metastatic Pancreatic Adenocarcinoma: A Randomized Phase III PRODIGE 65-UCGI 36-GEMPAX UNICANCER Study
C. De La Fouchardière et al, JCO 2024
Cette étude est un essai clinique de phase III ouvert et randomisé, conçu pour évaluer l'efficacité et la tolérance de l'association de la Gemcitabine et du Paclitaxel (GEMPAX) par rapport à la Gemcitabine seule, comme traitement de deuxième ligne pour les patients atteints d'adénocarcinome du pancréas métastatique (mPDAC), ayant précédemment reçu du 5-Fluorouracile, de l'Oxaliplatine et de l'Irinotécan. En effet, ces trois dernières molécules sont utilisées en association en première ligne de traitement du mPDAC pour les patients en suffisamment bon état général. Rappelons que la Gemcitabine seule est actuellement le standard en deuxième ligne. La survie des patients reste très limitée, avec environ 5 % de survie à 5 ans.
Les patients présentant un mPDAC confirmé histologiquement ou cytologiquement ont été assignés de manière aléatoire (2:1) pour recevoir GEMPAX (Paclitaxel 80 mg/m2 + Gemcitabine 1 000 mg/m2 ; IV ; une fois les jours (J) 1, J8 et J15/bras A) ou Gemcitabine (bras B) seule à J1, J8 et J15. Un cycle était démarré tous les 28 jours (les patients avaient donc une semaine sans traitement), jusqu'à la progression ou apparition d'une toxicité intolérable ou sur décision du patient. Le critère de jugement principal était la survie globale (OS). Les critères de jugement secondaires comprenaient la survie sans progression (PFS), le taux de réponse objective (ORR), la qualité de vie et la sécurité de l'association des chimiothérapies en termes de toxicité.
Au total, 211 patients (âge médian, 64 [30-86] ans ; 62 % d'hommes) ont été inclus. Après un suivi médian de l'étude pour les patients en vie de 13,4 versus 13,8 mois dans le bras A par rapport au bras B, la survie médiane (IC à 95 %) était de 6,4 (5,2 à 7,4) versus 5,9 mois (4,6 à 6,9 ; Hazard Ratio [HR], 0,87 [0,63 à 1,20] ; p = 0,4095), la PFS médiane était de 3,1 (2,2 à 4,3) versus 2,0 mois (1,9 à 2,3 ; HR, 0,64 [0,47 à 0,89] ; p = 0,0067), et le ORR était de 17,1 % (11,3 à 24,4) versus 4,2 % (0,9 à 11,9 ; P = 0,008) dans le bras A versus le bras B, respectivement. Dans l'ensemble, 16,7 % des patients dans le bras A et 2,9 % dans le bras B ont arrêté leur traitement en raison d'événements indésirables (AE). Un AE de grade 5 (correspondant à un décès) associé à l'association GEMPAX a été signalé dans le bras A (détresse respiratoire aiguë). À souligner également que 58,0 % vs 27,1 % des patients ont présenté des AE liés au traitement de grade supérieure ou égale 3 dans le bras A versus le bras B (15,2 % vs 4,3 % : anémie ; 15,9 % vs 15,7 % : neutropénie ; 19,6 % vs 4,3 % : thrombopénie ; 10,1 % vs 2,9 % : asthénie ; et 12,3 % vs 0,0 % : neuropathie).
Cette étude est donc négative sur son critère de jugement principal qui était la survie globale. Cependant, l'association de chimiothérapies GEMPAX, par rapport à la Gemcitabine seule chez les patients atteints de mPDAC en deuxième ligne, améliore significativement à la fois la PFS et le ORR. La toxicité rapportée dans cette étude est très accrue dans le groupe GEMPAX. Ces arguments sont en défaveur de considérer ce traitement comme un nouveau standard de seconde ligne pour la prise en charge du mPDAC.
CARv3-TEAM-E T Cells in Recurrent Glioblastoma
B.D. Choi et al, NEJM 2024
Cet article rapporte trois cas participants atteints de glioblastome en progression après une première ligne de traitement et qui ont été traités avec des lymphocytes T CARv3-TEAM-E. Il s'agit des trois premiers cas de patients aillant reçus des lymphocytes T à récepteur antigénique chimérique (CAR-T) conçus pour cibler l'antigène spécifique de tumeur EGFR variant 3 (EGFRv3), ainsi que la protéine EGFR de type sauvage, par le biais de la sécrétion d'une molécule d'anticorps engageant les lymphocytes T (TEAM). Le glioblastome est la tumeur cérébrale primitive la plus agressive, avec un pronostic à la rechute extrêmement sombre, sans options de traitement efficaces. Les CAR-T représentent une approche prometteuse contre le cancer en raison de leur efficacité prouvée contre de nombreux cancers hématologiques, pour lesquels ils sont devenus le traitement standard. Cependant, l'utilisation des CAR-T dans les tumeurs solides telles que les glioblastomes reste limitée jusqu'à présent, principalement en raison de la difficulté à cibler un seul antigène dans une maladie hétérogène avec également des mécanismes d'immunosuppression associés au microenvironnement tumoral.
Un précédent essai clinique en 2017 avait évalué un CAR-T-EGFRv3 sans activité TEAM. La tolérance et sécurité de ce CAR-T était bonne mais aucune réponse radiographique n'avait été observée, et les cellules tumorales à la récidive exprimaient la protéine EGFR de type « sauvage » avec une infiltration intra-tumorale importante par des lymphocytes T régulateurs suppressifs. C'est pour surmonter ces résistances que les chercheurs ont développé ce CARv3-TEAM-E qui cible donc, répétons-le, l'EGFRvIII à travers un CAR de deuxième génération tout en sécrétant des molécules d'anticorps engageant les lymphocytes T contre l'EGFR de type « sauvage », qui n'est pas exprimé dans le cerveau normal mais est presque toujours exprimé dans le glioblastome.
Concernant les résultats, le traitement n'a pas entraîné d'événements indésirables sévères (grade 3 ou plus), ni d'effets toxiques limitant la dose. Une régression tumorale radiographique a été spectaculaire et rapide, survenant dans les jours suivant la perfusion intraventriculaire cérébrale. Cependant, les réponses étaient transitoires avec une rechute pour les 3 patients à 72 jours, 150 jours et un mois respectivement. Malgré une réponse rapide radiologique ce nouveau CAR-T ne semble pas persister suffisamment dans le temps. Les auteurs soulignent aussi la perte de l'EGFRv3 avec ce traitement qui est un mécanisme de résistance. Nous remarquons que dans ce schéma de traitement, que les patients ne recevaient pas de lympho-déplétion, habituellement réalisée avant l'injection d'un CAR-T. C'est donc une étude très prometteuse avec un CAR-T original, ayant un bon profil de tolérance, mais des recherches sont à poursuivre pour permettre d'obtenir une durabilité de la réponse.
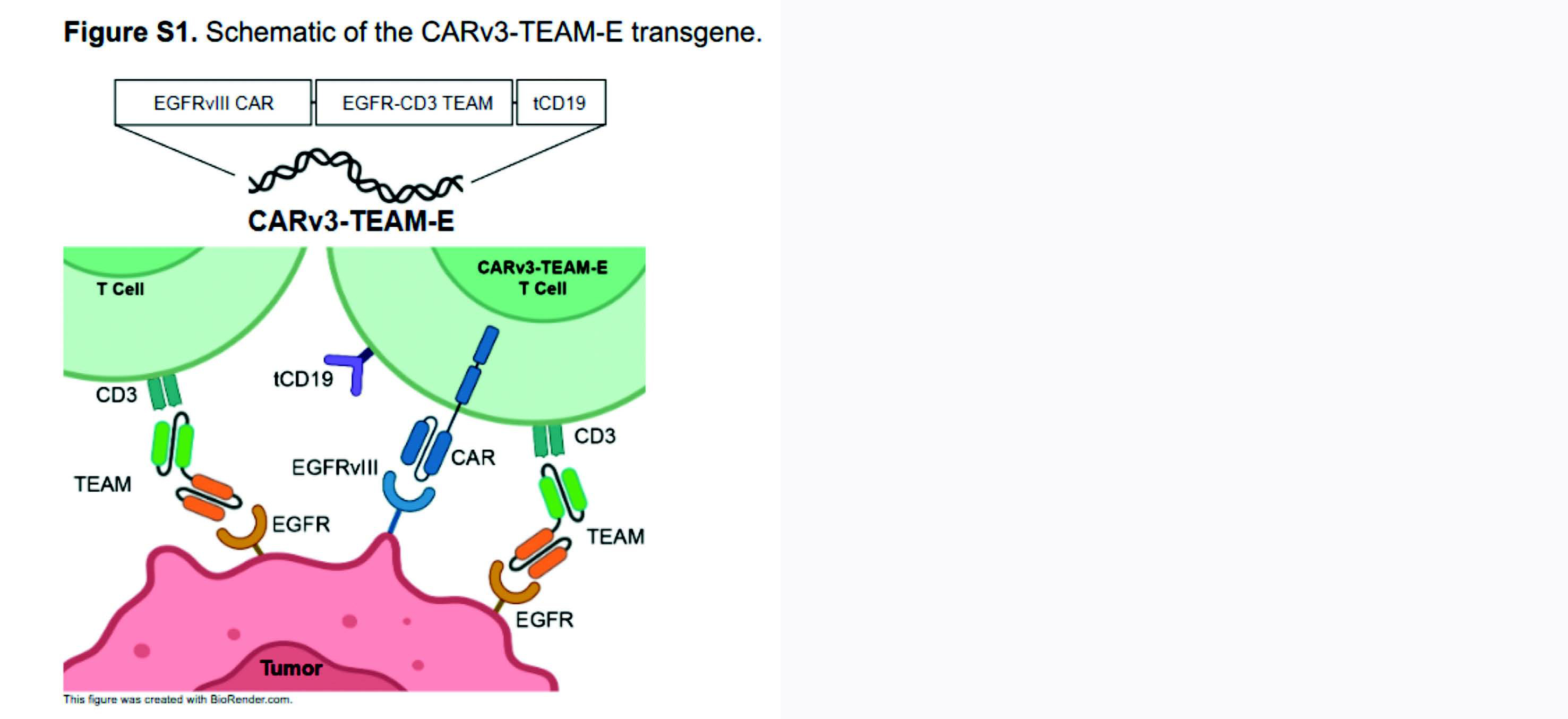
Mode de fonctionnement du CARv3-TEAM-E

Rédigé par Paul MATTE