


DNA methylation profiling of myelodysplastic syndromes and clinical response to azacitidine: a multicentre retrospective study
Aleix Noguera-Castells, Ignacio Campillo-Marcos, Veronica Davalos et al. British Journal of Haematology, 2024
Introduction
Les syndromes myélodysplasiques (SMD) représentent un groupe hétérogène de maladies, touchant essentiellement les sujets âgés. Ils sont caractérisés par une hématopoïèse inefficace aboutissant à des cytopénies sanguines et un risque accru de progression vers une leucémie aiguë myéloïde (LAM)1-3.
Les analyses de séquençage du génome entier ont identifié des mutations dans plusieurs gènes régulateurs de la méthylation de l'ADN, tels que TET2, DNMT3A, IDH1 et IDH21-3. De plus, les études épigénétiques du SMD ont montré l'existence d'un profil de méthylation aberrant chez les patients en comparaison aux sujets sains4, donnant un rationnel à l'utilisation d'agents hypométhylants (HMA) comme l'azacitidine en traitement des SMD de haut risque.
Une des limites de ce type de traitement est le faible taux de réponse qui est obtenue chez seulement 40 à 50 % des patients atteints de SMD de haut risque1-3. Bien que plusieurs biomarqueurs potentiels de réponse à l'azacitidine dans les SMD aient été proposés, un profil global et complet du méthylome de l'ADN des échantillons de SMD traités par HMA avant et après le traitement n'a pas été rapporté.
Il s'agit d'une étude descriptive d'épigénétique visant à comparer les profils de méthylation de l'ADN des patients atteints de syndromes myélodysplasiques (SMD) avant et après traitement par des agents hypométhylants (HMA), en fonction de leur réponse au traitement. L'objectif secondaire de cette étude est d'identifier une signature de méthylation de l'ADN qui serait capable de prédire l'efficacité du traitement, et ce, dès le diagnostic.
Matériels et méthodes
L'étude inclut 43 patients atteints de syndromes myélodysplasiques (SMD), avec 86 échantillons appariés de moelle osseuse recueillis avant et après traitement par azacitidine. Les profils de méthylation de l'ADN ont été générés à l'aide de la puce Infinium HumanMethyla tionEPIC (EPIC/850k). Les scores de méthylation (β et M-valeurs) ont été obtenus après un contrôle de qualité, une normalisation et un filtrage des fichiers bruts (IDATs)5. Le score global de la méthylation a été évalué en calculant le nombre de sites hyperméthylés. Les positions CpG différentiellement méthylées (DMPs) ont été identifiées en calculant les différences de valeurs moyennes de β entre les groupes et en ajustant un modèle de régression linéaire avec limma, en tenant compte de l'effet de lot comme covariable.
Un modèle de classification a été entraîné avec les DMPs en utilisant un algorithme de forêt aléatoire. La performance du modèle a été évaluée par la courbe ROC (receiver operating characteristic). Les p-va lues pour les variables clinico-pathologiques ont été calculées en utilisant le test exact de Fisher ou le test du chi-carré pour les variables dichotomiques ou catégorielles. Les comparaisons de méthylation globale entre les groupes ont été effectuées avec le test bilatéral de Mann-Whitney-Wilcoxon. La signification statistique était définie par une p-value inférieur à 0,05.
Résumé des résultats et discussion
La cohorte était composée majoritairement de patients âgés (plus de 70 ans pour 66,7 % des patients), les SMD étaient principalement classés en haut risque à très haut risque selon l'IPSS-R (76,7 %) et les patients recevaient en médiane six cycles de traitement.
Le taux de réponse à l'azacitidine était de 58.1 %, ce qui concorde avec les résultats d'autres études1-3 et la durée médiane de la réponse était de 10 mois.
Figure 1 : Contenu global de la méthylation de l'ADN
L'étude a d'abord déterminé le contenu global de la méthylation de l'ADN en calculant le nombre de sites CpG hyperméthylés (β-valeur Supérieur 0,66) pour chaque patient et chaque condition (pré et post-traitement).
Comme prévu, l'agent hypométhylant (HMA) a induit une réduction globale de la méthylation de l'ADN dans les échantillons post-traitement par rapport au groupe pré-traitement, comme en témoigne la diminution significative du nombre moyen de CpG hyperméthylés. Cette diminution de la méthylation a été observée dans tous les loci génomiques classés par densité et contexte CpG, tels que les zones riches en CpG (îlots CpG), les loci voisins des îlots CpG (étagères et rives CpG) et les régions pauvres en CpG (open sea).
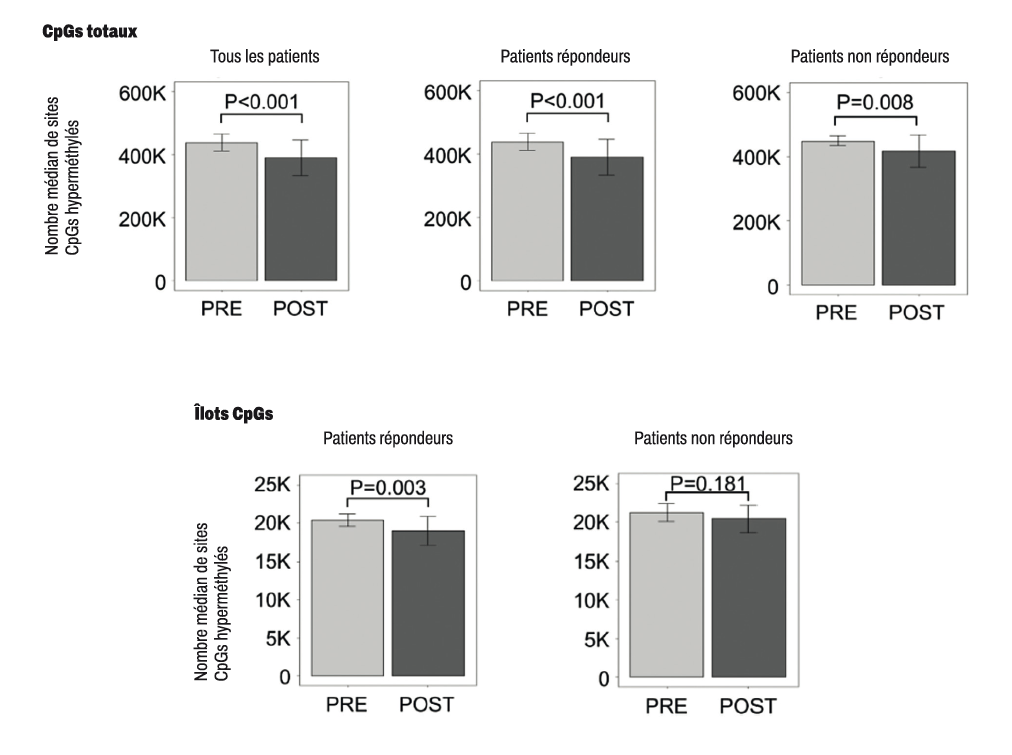
L'effet hypométhylant était globalement plus important chez les répondeurs aux HMA (p inférieur à 0,001) que chez les non-répondeurs (p-valeur = 0,008). Les patients répondeurs aux HMA ont montré une réduction significative des sites hyperméthylés situés aux îlots CpG (p = 0,003), cette réduction n'est pas observée chez les patients SMD réfractaires au traitement à l'azacitidine (p-valeur = 0,181). Pour les autres sites CpG, aucune différence n'a été observée entre répondeurs et non-répondeurs, les deux groupes montrant des événements significatifs d'hypométhylation suite à l'utilisation de l'azacitidine (p-valeur inférieur à 0,05).
Sites CpG individuels
Quatorze loci CpG ont été identifiés comme étant hypométhylés uniquement dans les échantillons post-traitement HMA des patients répondeurs. L'un des gènes associés à ces sites CpG identifiés, phosphoinositide-phospholipase C beta1 (PLCB1), a déjà été décrit7-8 comme réactivé lors de l'hypométhylation de l'ADN chez les patients SMD répondant au traitement HMA .
Analyse différentielle de la méthylation de l'ADN
Pour optimiser l'intérêt clinique, une analyse différentielle de la méthylation de l'ADN a été réalisée entre les répondeurs et les nonrépondeurs, en se concentrant uniquement sur les échantillons avant le traitement par azacitidine. Ces échantillons ont été divisés en cohortes d'exploration (n = 31) et de validation (n = 12). Aucune différence significative entre les deux cohortes n'a été trouvée concernant les variantes clinicopathologiques.
Signature EPIAZA
L'analyse de la cohorte d'exploration a révélé 39 sites CpG avec des différences significatives de niveaux de méthylation entre répondeurs et non-répondeurs (delta β-valeur moyenne Supérieur à|0,2| et p-valeur ajustée Inférieur à 0,05). Parmi les gènes liés à ces sites CpG, SLC35D2 et SLC22B5 appartiennent à la super-famille des transporteurs solutés précédemment décrits comme associés à la réponse à l'azacitidine9-10. Le gène lactate déshydrogénase C (LDHC), hyperméthylé chez les répondeurs HMA a été étudié plus en détail. Le statut de méthylation de ces quatre CpG permettrait à lui seul de classer les patients selon la réponse à l'azacitidine dans les échantillons pré-traitement.
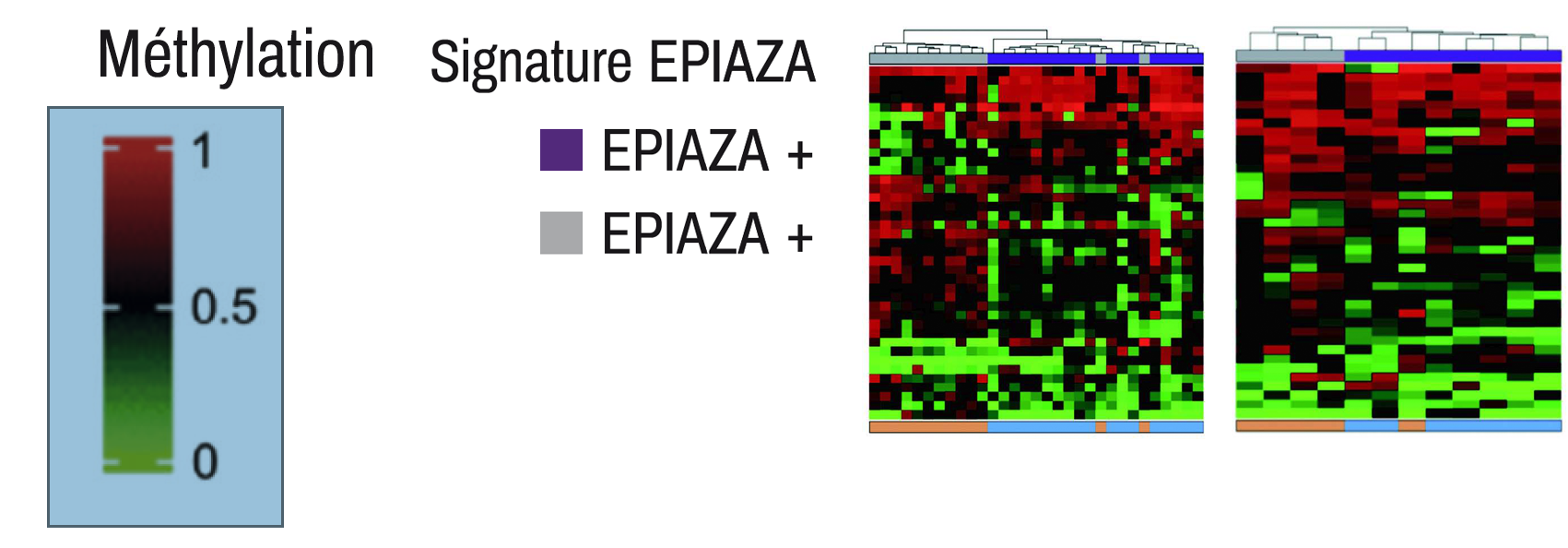
Cohorte exploratoire Cohorte de validation
Figure 3 : Performance de la signature EPIAZA
La signature EPIAZA a classé les patients MDS selon leur réponse clinique à l'azacitidine avec une précision de 91,7 % dans la cohorte de validation (IC 95 % = 62 %–99 % ; K = 0,82), avec une sensibilité de 80 % et une spécificité de 100 %. La courbe ROC a montré une AUC de 0,89. La signature EPIAZA a également prédit la réponse clinique à l'azacitidine pour les pourcentages faibles et élevés de blastes avec une grande précision et spécificité. Pour les patients à risque élevé et faible selon la classification IPSS-R, 95,24 % des cas (40 sur 42) étaient IPSS-R Supérieur à 3,5 %, et la signature EPIAZA était associée à la réponse HMA avec une précision de 98 % (IC 95 % = 87 %–99 %).
Conclusion
Cette étude confirme que les agents hypométhylants (HMA) induisent des changements globaux de méthylation de l'ADN chez les patients atteints de syndromes myélodysplasiques (SMD) et a identifié des signatures épigénomiques globales ainsi que des sites CpG spécifiques pouvant prédire la réponse clinique à ces agents déméthylants. Les résultats montrent que malgré un remodelage profond du paysage de la méthylation de l'ADN, les patients répondeurs présentent des événements d'hypométhylation préférentiels dans les régions régulatrices des îlots CpG. D'un point de vue clinique, l'analyse du méthylome de l'ADN avant traitement fournit déjà des indices sur l'évolution de la maladie. En ce sens, la signature EPIAZA obtenue pourrait être un outil précieux pour anticiper une réponse à la thérapie HMA chez les patients atteints de SMD de haut risque.

Abdessamia GANDOUL
Interne en Hématologie
CHU de Grenoble
Références
1. Platzbecker, U., Kubasch, A. S., Homer-Bouthiette, C., Prebet, T. Current challenges and unmet medical needs in myelodysplastic syndromes. Leukemia, 2021; 35(8): 2182–98.
2. Li, H., Hu, F., Gale, R. P., Sekeres, M. A., Liang, Y. Myelodysplastic syndromes. Nat Rev Dis Primers, 2022; 8(1): 74.
3. Rotter, L. K., Shimony, S., Ling, K., Chen, E., Shallis, R. M., Zeidan, A. M., et al. Epidemiology and pathogenesis of myelodysplastic syndrome. Cancer J., 2023; 29(3): 111–21.
4. Figueroa, M. E., Skrabanek, L., Li, Y., Jiemjit, A., Fandy, T. E., Paietta, E., et al. MDS and secondary AML display unique patterns and abundance of aberrant DNA methylation. Blood, 2009; 114(16): 3448–58.
5. Moran, S., Arribas, C., Esteller, M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics, 2016; 8(3): 389–99.
6. Davalos, V., Esteller, M. Cancer epigenetics in clinical practice. CA Cancer J Clin., 2023; 73(4): 376–424.
7. Cocco, L., Finelli, C., Mongiorgi, S., Clissa, C., Russo, D., Bosi, C., et al. An increased expression of PI-PLCβ1 is associated with myeloid differentiation and a longer response to azacitidine in myelodysplastic syndromes. J Leukoc Biol., 2015; 98(5): 769–80.
8. Follo, M. Y., Finelli, C., Mongiorgi, S., Clissa, C., Bosi, C., Testoni, N., et al. Reduction of phosphoinositide-phospholipase C beta1 methylation predicts the responsiveness to azacitidine in high-risk MDS. Proc Natl Acad Sci USA., 2009; 106(39): 16811–16.
9. Damaraju, V. L., Mowles, D., Yao, S., Ng, A., Young, J. D., Cass, C. E., et al. Role of human nucleoside transporters in the uptake and cytotoxicity of azacitidine and decitabine. Nucleosides Nucleotides Nucleic Acids., 2012; 31(3): 236–55.
10. Monika, B. M., Merkerova, M. D., Votavova, H., Valka, J., Vesela, J., Pejsova, B., et al. Up-regulation of ribosomal genes is associated with a poor response to azacitidine in myelodysplasia and related neoplasms. Int J Hematol., 2016; 104(5): 566–73.
11. Iorio, F., Knijnenburg, T. A., Vis, D. J., Bignell, G. R., Menden, M. P., Schubert, M., et al. A landscape of pharmacogenomic interactions in cancer. Cell., 2016; 166(3): 740–54.
12. Bonny, C., Goldberg, E. The CpG-rich promoter of human LDH-C is differentially methylated in expressing and nonexpressing tissues. Dev Genet., 1995; 16(2): 210–7.
13. Triozzi, P. L., Stirling, E. R., Song, Q., Westwood, B., Kooshki, M., Forbes, M. E., et al. Circulating immune bioenergetic, metabolic, and genetic signatures predict melanoma patients' response to anti-PD-1 immune checkpoint blockade. Clin Cancer Res., 2022; 28(6): 1192–202.
14. DiNardo, C. D., Jonas, B. A., Pullarkat, V., Thirman, M. J., Garcia, J. S., Wei, A. H., et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med., 2020; 383(7): 617–29.
15. Bazinet, A., Darbaniyan, F., Jabbour, E., Montalban-Bravo, G., Ohanian, M., Chien, K., et al. Azacitidine plus venetoclax in patients with high-risk myelodysplastic syndromes or chronic myelomonocytic leukaemia: phase 1 results of a single-centre, dose-escalation, dose-expansion, phase 1–2 study. Lancet Haematol., 2022; 9(10): e756–e765.