


Longtemps considérée comme une maladie incurable, l'amyotrophie spinale (SMA) connaît depuis quelques années une véritable révolution thérapeutique. Des traitements innovants changent radicalement le pronostic et les perspectives de rééducation de ces patients, nous allons les détailler ci-dessous.
La SMA est une maladie neuromusculaire génétique, par mutation du gène SMN1. Cette mutation entraîne un déficit de production de la protéine SMN (survival motor neuron), essentielle à la survie des motoneurones alpha de la corne antérieure de la moelle épinière. La sévérité de la maladie dépend notamment du nombre de copies du gène SMN2, un gène "de secours" qui produit une petite quantité de protéine SMN fonctionnelle. Plus le nombre de copies de SMN2 est élevé, plus le phénotype est modéré. La classification est détaillée dans le tableau 1 (1).
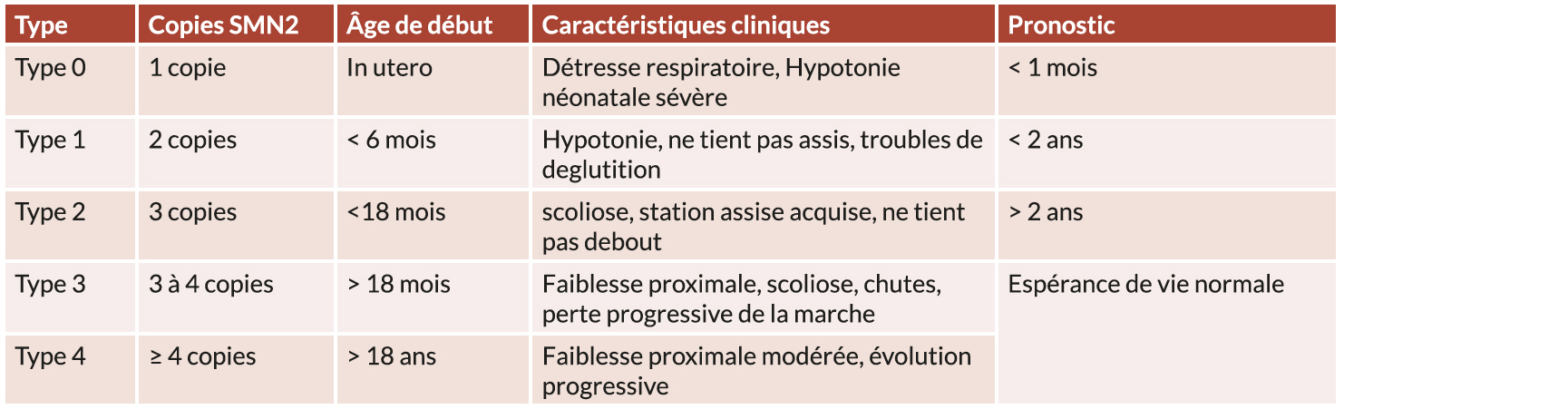
Tableau 1 : Classification actuelle de la SMA
Nusinersen (Spinraza®) (2)
Il s'agit du premier traitement découvert pour la SMA. C'est un oligonucléotide antisens administré par voie intrathécale. Il agit sur l'épissage alternatif du gène SMN2 pour augmenter la production de la protéine SMN fonctionnelle. Il est administré tous les 4 mois par injection intrathécale, car les oligonucléotides ne passent pas la barrière hémato-encéphalique.
Onasemnogene abeparvovec (Zolgensma®) (3)
Thérapie génique révolutionnaire autorisée depuis 2020 en ATU, Zolgensma délivre une copie fonctionnelle du gène SMN1 via un vecteur viral (AAV9). Une seule injection intraveineuse, en général avant 2 ans, peut suffire à stopper la progression de la maladie, mais ne permet pas de récupérer les motoneurones perdus. Il est indiqué dans la SMA de type 1 et 2 ou en présymptomatique chez les patients ayant jusqu'à 3 copies de SMN2.
Risdiplam (Evrysdi®) (4)
Commercialisé plus récemment, Risdiplam est un traitement oral, quotidien, modificateur d'épissage du gène SMN2. Il permet une meilleure observance, notamment chez les patients plus âgés ou inéligibles au Spinraza.
Les résultats
Une cohorte nationale française a publié en décembre 2024 les premiers résultats en vie réelle, trois ans après la commercialisation du Zolgensma® (5). L'étude a inclus 29 patients atteints de SMA de type 1 traités par thérapie génique. Parmi eux, deux sont décédés peu après l'injection : un à la suite d'une microangiopathie thrombotique, l'autre d'une détresse respiratoire.
Sur les 29 patients, des données de suivi à 2 ans étaient disponibles pour 17 d'entre eux. À deux ans post-injection :
• 100 % des enfants tenaient leur tête ;
• 15 sur 17 pouvaient s'asseoir seuls pendant 30 secondes ;
• 12 sur 17 pouvaient se maintenir brièvement debout avec appui.
Les fonctions respiratoires et alimentaires étaient globalement préservées, bien que la majorité ait nécessité un corset rachidien (15/17). Certains patients ont eu recours à une ventilation nocturne (3/17) ou à une gastrostomie (2/17).
Selon les données du laboratoire Novartis, ces acquisitions motrices sont maintenues jusqu'à 7,5 ans après traitement, ce qui témoigne de la durabilité de la réponse.
Ces résultats sont d'autant plus remarquables que, rappelons-le, les patients atteints de SMA type 1 avaient auparavant une espérance de vie inférieure à 2 ans, en l'absence de traitement.
Quelles perspectives pour ces nouveaux traitements ?
L'essai SPRINT, réalisé chez des nourrissons présymptomatiques atteints de SMA de type 1, a confirmé qu'un traitement précoce est nettement plus efficace lorsqu'il est administré avant l'apparition des premiers symptômes (6). Cela souligne l'importance capitale du dépistage néonatal.
En France, le projet DEPISMA a démontré la faisabilité du dépistage systématique de la SMA à la naissance (7).
La généralisation du dépistage néonatal de la SMA est ainsi prévue à partir du 1er septembre 2025.
Aussi, avec le traitement présymptomatique, la clinique des patients change et cela implique de repenser la classification actuelle qui se base sur l'âge de début des symptômes et le développement psychomoteur de l'enfant (8).
Quelle implication pour nous, internes en MPR ?
Ces avancées thérapeutiques bouleversent l'histoire naturelle de la SMA et appellent à une réinvention du suivi en MPR. Il nous reviendra de mettre en place un suivi à long terme et d'adapter les objectifs de rééducation à l'évolution motrice de ces enfants.
Références
1. https://www.mda.org/disease/spinal-muscular-atrophy/types
2. https://afm-telethon.com/fr/vivre-avec-la-maladie/mon-parcours-de-soins/les-medicaments/le-spinrazar-dans-la-sma
3. https://www.afm-telethon.fr/fr/vivre-avec-la-maladie/mon-parcours-de-soins/les-medicaments/le-zolgensmar-dans-la-sma
4. https://afm-telethon.com/fr/vivre-avec-la-maladie/mon-parcours-de-soins/les-medicaments/levrysdir-dans-la-sma
5. Desguerre, I., et al. Real-world multidisciplinary outcomes of onasemnogene abeparvovec monotherapy in patients with spinal muscular atrophy type 1: experience of the French cohort in the first three years of treatment. Orphanet J Rare Dis 19, 344 (2024). https://doi.org/10.1186/s13023-024-03326-3
6. Strauss KA, et al. Onasemnogene abeparvovec for presymptomatic infants with two copies of SMN2 at risk for spinal muscular atrophy type 1: the Phase III SPR1NT trial. Nat Med. 2022 Jul;28(7):1381-1389. doi: 10.1038/s41591-022- 01866-4. Epub 2022 Jun 17. PMID: 35715566; PMCID: PMC9205281.
7. Didier Lacombe et al : Dépistage génétique néonatal : à propos du programme pilote sur l'amyotrophie spinale (DEPISMA),Bulletin de l'Académie Nationale de Médecine, Volume 208, Issue 1, 2024. https://doi.org/10.1016/j.banm.2023.09.019
8. Varone Antonio , et al : Spinal muscular atrophy in the era of newborn screening: how the classification could change. Frontiers in Neurology ; Volume 16 – 2025. DOI=10.3389/fneur.2025.1542396.
Maria ZAKHEM