


Il existe plus de 300 maladies neuromusculaires, qui se manifestent par une atteinte musculaire transitoire ou permanente qui peut apparaître à tout âge de la vie et s'aggraver plus ou moins rapidement.
Les symptômes sont :
• Faiblesse musculaire : elle se manifeste différemment selon l'âge d'apparition (hypotonie du nourrisson, retard de développement psychomoteur, chutes, difficultés à marcher ou monter les escaliers). Elle se manifeste également différemment selon les muscles atteints : déficit des muscles proximaux dans la dystrophie musculaire de Duchenne, l'amyotrophie spinale ou les myopathies des ceintures ; déficit des muscles distaux (mains, pieds/chevilles) comme dans la maladie de Charcot-Marie-Tooth ou la dystrophie myotonique de Steinert ; déficit des muscles du visage qui peuvent aussi être concernés (dans la myopathie facio-scapulo-humérale, la dystrophie myotonique de Steinert ou les myopathies congénitales) ; atteinte des muscles de la déglutition dans la dystrophie musculaire oculopharyngée.
• Les complications de cette faiblesse musculaire sont notamment les rétractions musculo-tendineuses et les troubles de la statique rachidienne (cyphose et/ou scoliose).
• Difficultés cognitives : dans les dystrophies musculaires de Duchenne et de Steinert.
• Autres symptômes possibles : difficulté au relâchement musculaire (myotonie), hypermobilité articulaire, troubles cardiaques, atteinte ophtalmologique et/ou de l'audition...
Localisation de l'atteinte
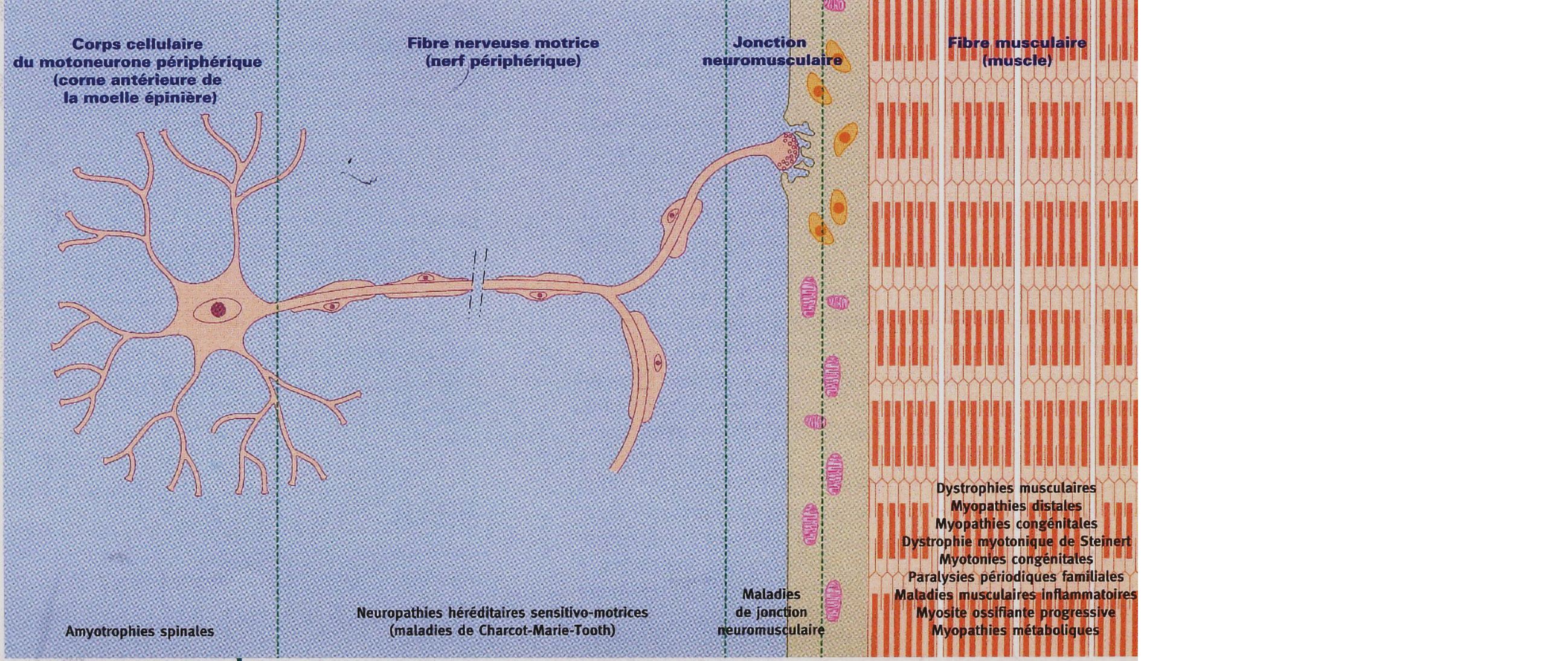
Figure 1 : Atteinte de l'unité motrice
Les différentes maladies neuro-musculaires peuvent être classées selon la localisation de l'atteinte au niveau de l'unité motrice :
Atteinte du motoneurone
Amyotrophie spinale infantile, sclérose latérale amyotrophique…
L'amyotrophie spinale est une maladie génétique à transmission autosomique récessive causée par un défi- cit en protéine SMN (Survival Motor Neuron), qui cause une dégénérescence des neurones moteurs de la corne antérieure (et des noyaux bulbaires). L'incidence est de 1/6 000. Elle se caractérise par un déficit musculaire à prédominance proximale et symétrique, une abolition des réflexes ostéo-tendineux, des fasciculations linguales.
La sclérose latérale amyotrophique (SLA) est due à la mort progressive des motoneurones, qu'ils soient centraux ou périphériques. L'incidence en France est de 2,5/100 000 habitants. Elle est d'origine multifactorielle, environnementale (certains facteurs sont suspectés tels que le tabac, le sport de haut niveau, l'expo sition à des pesticides, à des métaux lourds ou encore à une toxine présente dans certaines algues) et génétique (10 % de formes familiales). Elle apparaît le plus souvent entre 50 et 70 ans, parfois plus précocement dans les formes familiales. Elle prend des formes différentes selon la nature de l'atteinte initiale : 30 % de formes à début bulbaire dont les premières manifestations sont les difficultés à articuler ou à déglutir, et 70 % de formes à début spinal, les premières manifestations pouvant être une faiblesse et une gêne au niveau d'un bras, d'une jambe ou d'une main. Il s'agit d'une maladie au pronostic sombre, dont l'issue est fatale après 3 à 5 ans d'évolution en moyenne (du fait d'une atteinte des muscles respiratoires principalement).
Neuropathies sensitivomotrices héréditaires
Maladie de Charcot-Marie-Tooth
Concernant la maladie de Charcot-Marie-Tooth, il s'agit d'une atteinte des nerfs périphériques (sensitifs et/ou moteurs) concernant soit la gaine de myéline soit l'axone. Elle est fréquente (1/2500), à transmission autosomique dominante ou autosomique récessive ou dominante liée à l'X. Elle débute majoritairement dans l'enfance (50 %) ou chez l'adulte jeune, et se caractérise par des troubles de la marche (fatigabilité, marche précautionneuse), un déficit des releveurs de pied, une amyotrophie des mollets, des pieds creux avec griffe des orteils, des réflexes ostéo-tendineux abolis, des troubles de la sensibilité superficielle et profonde, et des douleurs. L'évolution est variable, avec parfois une aggravation de la déformation des pieds, la survenue d'une atteinte proximale ou d'une atteinte des mains avec déficit de la musculature intrinsèque.
Atteinte de la jonction neuromusculaire
Myasthénie acquise, myasthénie congénitale…
Cette fois-ci, la myasthénie acquise est une maladie non génétique de type auto-immune, avec des anticorps se fixant sur les récepteurs de l'acétylcholine au niveau de la jonction neuro-musculaire. L'incidence est de 5/100 000, Elle présente deux pics d'incidence : entre 20 et 40 ans chez la femme, après 60 ans chez l'homme. Les symptômes sont une fatigue et une faiblesse musculaire s'aggravant à l'effort et en fin de journée, évoluant par poussées avec des rémissions plus ou moins complètes. Il est important de connaître les signes de crise myasthénique car elle peut entrainer une détresse respiratoire grave.
La myasthénie congénitale quant à elle est une maladie génétique, à transmission le plus souvent autosomique récessive, dont les symptômes surviennent dès la naissance ou la petite enfance. L'atteinte est plus généralisée, touchant également les muscles respiratoires, et avec plus de déformations orthopédiques comme des scolioses. L'incidence est de 0,23/100 000.
Atteinte de la fibre musculaire
Dystrophies musculaires (dystrophie musculaire congénitale, dystrophinopathie de Duchenne ou de Becker, dystrophie musculaire des ceintures, dystrophie musculaire scapulo- humérale, dystrophie myotonique de Steinert…), myopathies congénitales (à bâtonnets, central core…), myopathies métaboliques (glycogénoses, mitochondriales, lipidoses…)
Concernant la dystrophie musculaire de Duchenne (DMD), il s'agit d'une pathologie génétique à transmission récessive liée à l'X, causant une altération dans le gène DMD localisé sur le chromosome X codant la dystrophine. Elle touche donc uniquement les garçons, avec une incidence de 1/3500. Le début des signes survient dans la petite enfance : retard d'acquisition mais surtout anomalies de la marche, difficultés pour se relever du sol (atteinte proximale), troubles statiques (hyperlordose, protrusion abdominale, équin, épaules en arrière), et pseudohypertrophie des mollets, accompagnés d'un déficit cognitif dans 30-40 % des cas. Cette pathologie évolue vers une aggravation progressive du déficit avec perte de la marche qui apparaît entre 10 et 12 ans, une atteinte des membres supérieurs un peu plus tardive, des complications orthopédiques précoces (rétractions musculaires et déformations articulaires), des complications respiratoires (survenue vers 14-18 ans), une atteinte cardiaque (troubles du rythme, diminution de la fraction d'éjection du ventricule gauche), des troubles nutritionnels, et un décès prématuré.
La dystrophie musculaire de Becker est dix fois moins fréquente. Elle se caractérise par une altération sur la même localisation (réduction de la quantité ou anomalie de la dystrophine), et présente donc les mêmes signes mais d'apparition plus tardive (atteintes des ceintures, crampes à l'effort), sans déficit cognitif associé. La progression est plus lente, très variable en ce qui concerne l'âge de la perte de la marche (50 % avant 40 ans). Le risque porte sur l'atteinte cardiaque qui est potentiellement évolutive.
Concernant les dystrophies musculaires des ceintures (LGMD = Limb Girdle Muscular Dystrophy), il s'agit d'un groupe hétérogène de pathologies à transmission autosomique dominante ou autosomique récessive, avec un âge de début très variable, et une incidence de 5 à 6/1 000 000 personnes. Les symptômes sont une faiblesse avec atrophie des muscles des ceintures scapulaire et pelvienne, d'évolution parfois sévère avec perte de la marche, parfois plus modérée avec une simple fatigabilité. Les complications respiratoires sont rares et non évolutives. Il existe de rares mais sévères atteintes cardiaques, et pas d'atteinte intellectuelle.
La dystrophie myotonique de Steinert vous sera présentée page 22.
La dystrophie musculaire facio-scapulo-humérale (dystrophie FSH, myopathie de Landouzy-Dejerine) : de transmission autosomique dominante (localisation de l'anomalie sur le chromosome 4), elle débute dans l'enfance, l'adolescence ou chez l'adulte jeune. Elle touche 2500 personnes en France. Elle se caractérise par une atteinte bilatérale mais asymétrique des muscles de la face (mimique peu expressive, difficultés pour souffler ou siffler, troubles du langage ou de la fermeture des yeux), mais pas d'atteinte des muscles oculomoteurs, de la langue, du pharynx, du larynx, des masséters et des temporaux. Il existe également un déficit de la ceinture scapulaire avec impossibilité d'élévation des bras, et parfois une atteinte des membres inférieurs (fessiers, releveurs), de la vision et de l'audition. Il n'y a pas d'atteinte respiratoire et cardiaque. L'évolution est très lente avec des périodes de stabilisation et une espérance de vie non modifiée, les patients présentent surtout une gêne fonctionnelle (instabilité à la marche).
Les myopathies congénitales sont des pathologies relativement fréquentes, dont le diagnostic est suspecté sur la notion d'hypotonie néonatale, un retard du développement moteur, une faiblesse et une gracilité musculaire diffuse. Elles sont peu ou non évolutives, la sévérité est fonction de l'âge d'apparition de la maladie. Nous pouvons retrouver la myopathie à bâtonnets, avec sa forme précoce présentant une hypotonie sévère, des déformations articulaires, une atteinte de la fonction respiratoire, des anomalies cardiaques et des troubles de la déglutition ; mais également des formes tardives avec des déformations des pieds et la survenue de scolioses. Nous pouvons également citer les myopathies à central cores qui sont les myopathies congénitales les plus fréquentes, la myopathie centronucléaire, la myopathie myotubulaire…
Concernant les myopathies métaboliques, nous allons en citer 3. Les glycogénoses musculaires (ex : Maladie de Pompe (type II), Maladie de Mac Ardle (Type V)) débutent à n'importe quel âge, et se traduisent par une fatigue musculaire, des myalgies à l'effort et une accumulation du glycogène non utilisé dans différents tissus de l'organisme (atteinte hépatique, cardiaque…). Les lipidoses musculaires sont dues à un déficit d'enzymes musculaires impliquées dans le métabolisme des lipides, et se manifestent lors de l'exercice ou du jeûne par une intolérance à l'effort avec des douleurs musculaires pendant ou après l'exercice, ainsi qu'une confusion mentale, une atteinte cardiaque, et un risque d'hypoglycémie. Les myopathies mitochondriales quant à elles ont des manifestations cliniques et une évolution très variables : chez l'enfant elles sont sévères avec une hypotonie, une atteinte cérébrale, hépatique, rénale et cardiaque ; chez l'adulte elles se présentent sous la forme d'une fatigabilité douloureuse à l'effort avec chute des paupières et ophtalmoplégie.
Myotonies congénitales
Elles sont caractérisées par une lenteur anormale du relâchement musculaire aggravée par le froid, améliorée par l'échauffement, non évolutive. L'incidence est de 1/100 000.
Ex : myotonie de Becker (AR) ou de Thomsen (AD) : mutations dans le gène : CLCN-1 (canal chlore).
Fibrodysplasie ossifiante progressive
Il s'agit d'une surexpression d'une protéine de l'os de transmission autosomique dominante ou sporadique, très rare (6/10 000 000). Elle débute dans l'enfance et évolue par poussées douloureuses qui apparaissent spontanément ou après un traumatisme même minime, suivies d'ossifications des muscles causant des limitations articulaires. On retrouve également des malformations congénitales des orteils (hallux valgus) et/ou du pouce (microdactylie).
Carte d'urgence : Carte personnelle de soins et d'information distribuée aux patients atteints de maladies rares pour améliorer la coordination de leurs soins notamment en cas d'urgence. Elles sont distribuées par les médecins spécialisés des centres maladies rares.

Spécimen de carte d'urgence pour maladie neuromusculaire, proposé par la filière FILNEMUS
Sources
• AFM téléthon
• Orphanet
• INSERM
Merci au Dr Sylvie RAGOT-MANDRY, médecin MPR à l'Hôpital d'enfants du CHRU de Nancy, de m'avoir fourni les cours nécessaires à la rédaction de cet article.
Dr Emma PETITJEANS