


Acromégalie infraclinique
Cas clinique 1 - Patiente de 43 ans aux antécédents de RCUH depuis plus de 10 ans (traitement par Entyvio, Pentaza), hernie discale, canal carpien bilatéral, abdominoplastie. G3P3, macrosomie fœtale 1ère grossesse, diabète gestationnel à la 3ème grossesse. Sous stérilet hormonal (aménorrhée).
Apparition brutale d'une surdité unilatérale faisant réaliser une IRM cérébrale complétée par une IRM hypophysaire : découverte d'adénome intra-sellaire de 8mm hyposignal T1, hypersignal T2 faiblement rehaussé après injection. Surdité de résolution spontanée en 24H sans étiologie retrouvée.
Quels signes d'acromégalie peut-on rechercher qui apparaîtraient en 1er ?
La temporalité d'apparition des signes est mal définie mais l'infiltration est présente dans 90 % des cas au diagnostic :
- Responsable de gonflement (signe de la bague, changement de pointure) avec parfois des fourmillements.
- Puis des modifications de faciès, des ronflements…
Le score d'activité de l'acromégalie « SAGIT » de 0 à 22 proposé par un consensus d'experts décrit les 4 signes d'activité principaux : céphalées, sueurs, arthralgies, gonflement. Certaines comorbidités sont présentes dès le diagnostic (DT2, HTA).
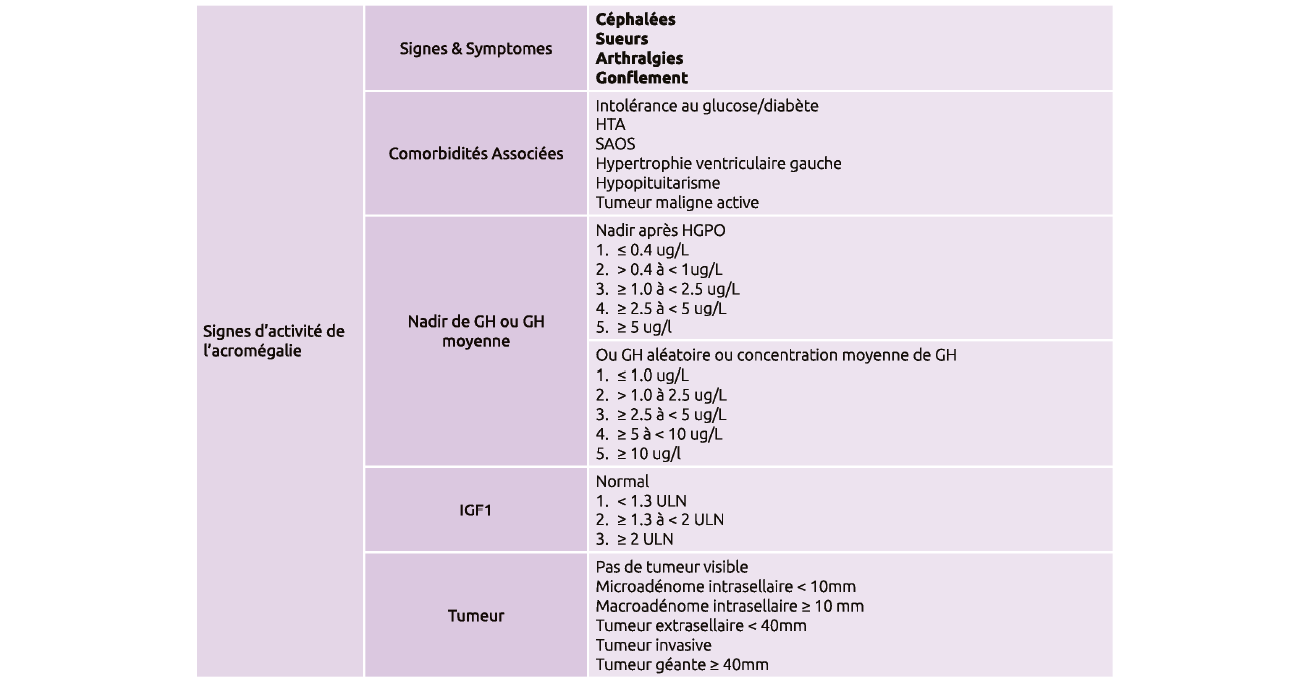
À noter chez cette patiente l'absence de symptômes, pas de troubles du sommeil, pas de signes d'acromégalie, d'hypercortisolisme ni de dysthyroïdie, pas de galactorrhée.
Quels dosages réaliser obligatoirement devant un incidentalome hypophysaire inférieur à 10mm ?
A. TSH, T4L
B. IGF1
C. Prolactine
D. Freinage minute du cortisol
E. LH, FSH, œstrogènes
Réponses : B et C (Consensus de la SFE sur les microadénomes hypophysaires de découverte fortuite) : limiter les dosages au strict nécessaire, soit IGF1 et prolactine + D ici le freinage minute était également réalisé du fait d'un IMC à 34 avec une TA à 160/100 mmHg.
Résultats : IGF1 élevée à 396 ng/ml (73–263), freinage minute et prolactine normales.
IGF1 élevée ne signifie pas toujours acromégalie !
- En pratique, rares sont les causes de ‘fausses augmentations' d'IGF1 : grossesse, puberté, hyperthyroïdie.
- Mais il y a une variabilité technique (6 trousses de dosages d'IGF1 différentes) et une variabilité individuelle (taux d'IGF1 différents d'une prise de sang à l'autre).
- On ne peut parler d'acromégalie que si l'IGF1 est supérieure à 1.3 x norme supérieure pour l'âge avec des signes cliniques (recommandations Pituitary Society) sinon, répéter le dosage d'IgGF1 + HGPO.
- Une concentration normale en IGF1 permet d'exclure l'acromégalie (PNDS HAS).
Pour confirmer l'acromégalie : IGF1 recontrôlée à 340 ng/ml et HGPO avec nadir de GH à 0,39 ng/ml, valeur maximale à
T0 à 0,81 ng/ml. Glycémie à jeun 1,12 g/L, glycémie à H2 1.69 g/L. HbA1c 6.2 %.
Ici on pourrait croire que son HGPO est normale sur le nadir de GH car inférieure à 0,4 μg/L mais la patiente ayant un IMC à 34, la GH partiellement freinée à 0,37 est pathologique en contexte d'obésité.
Les œstrogènes impactent peu la GH mais la sécrétion très impactée par l'IMC : le seuil de GH inferieur à 0,4 μg/L est abaissé à 0.2 μg/L en cas d'IMC supérieur à 25 kg/m2.
À noter que des sujets sains auront aussi une GH qui ne sera pas freinée lors de l'HGPO.
Quel diagnostic suspecter chez cette patiente ?
A. Acromégalie.
B. Micromégalie.
C. Adénome somatotrope silencieux.
D. Adénome somatotrope cliniquement silencieux.
Différents spectres des hypersécrétions de GH :
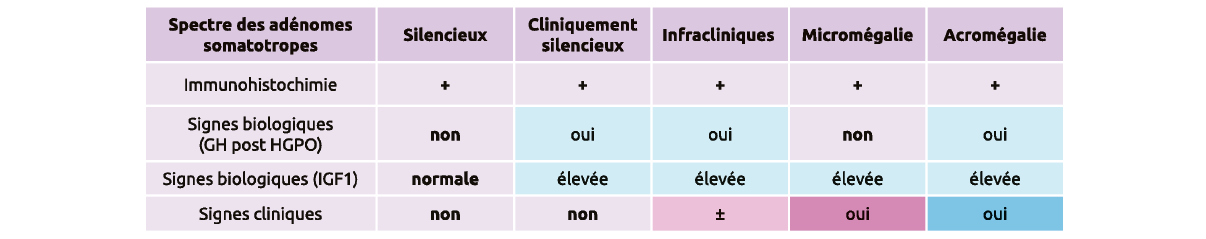
La micromégalie
Attention, malgré son nom qui peut être trompeur, la micromégalie n'est pas une hypersécrétion infraclinique !
Elle est caractérisée par :
- Une IGF 1 augmentée.
- Une GH normale ou peu élevée.
- Des signes cliniques d'acromégalie.
Les hypothèses physiopathologiques sont une sécrétion plus « tonique » de la GH (faible mais suffisante pour entraîner l'IGF1 élevée et les signes cliniques associés) ou un polymorphisme du récepteur de la GH qui entraînerait une sensibilité accrue ou un isoforme actif de la GH non dosée.
Les adénome somatotropes silencieux (ou acromégalie clinique silencieuse) sont découverts sur des incidentalomes. Des études retrouvaient une prévalence de 33 %, généralement des macroadénomes, plus fréquents chez la femme, âges variables mais sans signes d'acromégalie.
L'absence de signes cliniques pourrait s'expliquer par une découverte au stade précoce. Des modifications de faciès pourront être détectées précocément notamment via intelligence artificielle. D'autres hypothèses seraient une pauci sécrétion ou une sensibilité moindre du récepteur à la GH mais les donn.es de littérature sont discordantes.
Quelle prise en charge proposer pour les adénomes infracliniques ?
A. Surveillance simple.
B. Traitement médical seul par analogue de la somatostatine.
C. Prise en charge chirurgicale après préparation par analogue de la somatostatine.
D. Prise en charge chirurgicale sans traitement médical préalable.
Réponse : D, patiente en attente de chirurgie.
Il est difficile de prédire l'évolution clinique de ces patients : vont-ils développer une acromégalie patente ?
Une étude rétrospective de 53 patients ayant eu une chirurgie hypophysaire ou un incidentalome avec IGF1 augmentée (2 groupes suivis. 6 ans) et GH non augmentée, retrouvait plus de comorbidités chez les patients ayant des signes cliniques (=micromégalie) que chez les cliniquement silencieux. 3 patientes ayant des adénomes cliniquement silencieux suivies à 8 mois, 3 ans et 5 ans ont développé des signes d'acromégalie.
Les nouvelles recommandations 2025 retiennent une indication de chirurgie sans traitement préalable dans les acromégalies silencieuses ou infraclinique.
Ressources
Score d'activité SAGIT.
PNDS Acromégalies (HAS, 2021).
Melmed, S., di Filippo, L., Fleseriu, M. et al. Consensus on acromegaly therapeutic outcomes: an update. Nature Rev (2025).
Syndrome de cushing infraclinique
Patient de 51 ans, aux antécédents de carcinome épidermoïde de la cavité buccale, BPCO post-tabagique, RGO, cholecystéctomie, sous Inexium, Seresta, Formoair beta 2 stimulant sélectif.
Lors de l'évaluation de son carcinome épidermoïde, le scanner thoracique met en évidence un nodule surrénalien gauche unique de 33mm, densité spontanée 8 UH.
Il n'a pas de point d'appel cliniques, un IMC à 29 kg/m2, PA 127/81, FC 90 bpm.
Quels dosages systématiques réaliser devant cet incidentalome surrénalien ?
A. Freinage minute du cortisol.
B. Métaéphrines plasmatiques.
C. Aldostérone/rénine.
D. 17-OH-progestérone.
E. Androgénes surrénaliens.
Réponse : A (d'après les recommandations européennes de 2023 Fassnacht, EJE) Chez ce patient, freinage pathologique à 2.8 ug/dl.
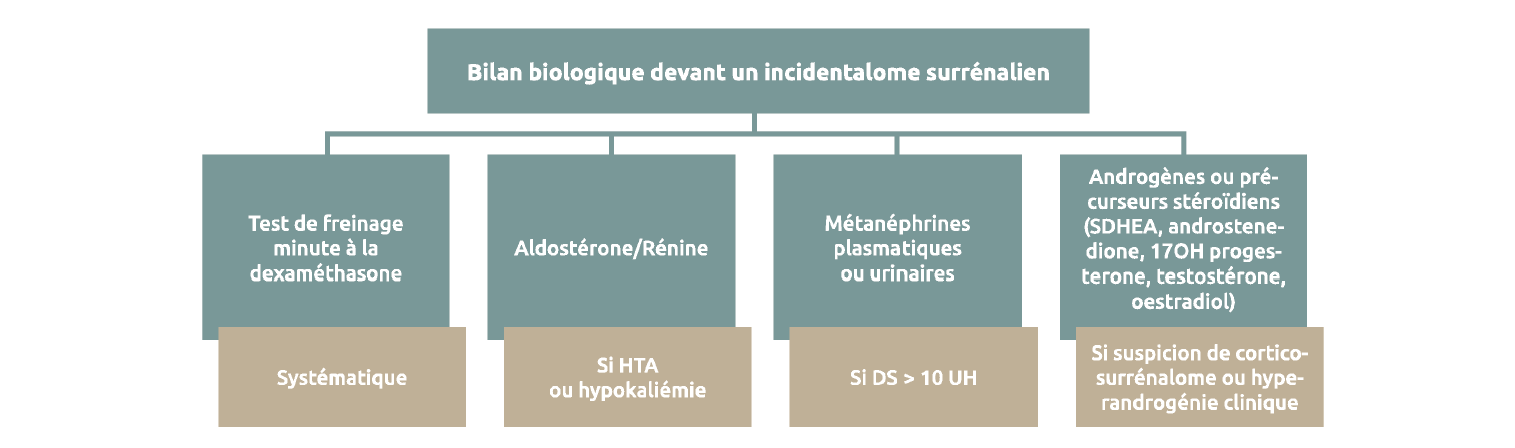
20 à 50 % des incidentalomes surrénaliens ont une sécrétion autonome de cortisol.
Un cortisol après freinage minute supérieur à 50 nmol/L ou supérieur à 1.8 μg /dl doit toujours faire considérer un MACS. La Sensibilité est de 99,2 % mais la spécificité est de 86 % : il est recommandé de répéter le test de freinage minute. Les limites du test de freinage sont :
- la contraception oestroprogestative ;
- la grossesse ;
- le métabolisme inter-individuel du CYP3A4.
A noter qu'il est possible de doser la dexaméthasonémie à l'hôpital.
Le patient est revu 2 ans après, présente une HTA traitée par IRBESARTAN 300mg et DILTIAZEM LP 100mg. Il a pris 9kg suite à un arrêt du tabac, soit un IMC à 35,7 kg/m2.
La réévaluation biologique retrouve un test de freinage pathologique à 1,9 et 5,1 μg/dl. L'ACTH est à 13 pg/ml à 8h, le cycle est conservé, le CLU est à 40 μg/24h.
Il n'a pas de signes de Cushing, mais des comorbidités : HTA, dyslipidémie, Obésité grade I, glycémie à jeun 1,1 g/L.
NB : il n'est pas nécessaire de réaliser un cycle de cortisol dans le MACS même si c'est fréquemment fait en hospitalier.
Une étude allemande sur les comorbidités du MACS montrait que plus de 35 % des patients classés MACS avaient au moins 2 symptômes cliniques de Cushing, alors qu'il y a des rares syndromes de Cushing avec seulement 1 signe clinique : on retrouve un chevauchement entre les 2.
Quelle prise en charge discuter chez ce patient ?
A. Une surveillance, le MACS n'a pas de conséquences cliniques.
B. Une surveillance car son test de freinage est inférieur à 5 μg /dl.
C. Une chirurgie car risque d'évolution vers un syndrome de Cushing.
D. Une chirurgie car il est hypertendu.
Réponses : D
Les MACS surrénaliens unilatéraux ont un risque faible voir inexistant de développer un syndrome de Cushing clinique, mais on retrouve une augmentation de la mortalité et de la morbidité de ces patients (risque relatif de diabète, HTA, dyslipidémie) avec un risque cardiovasculaire qui augmente en fonction du test de freinage, dès le seuil de supérieur à 50 nmol/L ou supérieur à 1.8 μg /dl.
Prise en charge du MACS
1. Test de freinage minute positif.
2. S'assurer qu'il ne s'agit pas d'un vrai syndrome de Cushing (clinique plus ou moins CLU et ACTH. 8h).
3. Une fois le MACS confirmé, rechercher la présence de comorbidités.
- Si comorbidités : s'assurer que ce n'est pas ACTH dépendant et considérer un traitement.
- Si pas de comorbidités : suivi à la recherche de comorbidités.
L'étude prospective randomisée contrôle CHIRACIC (Tabarin et al, Lancet EndocDiab, 2025) montre un bénéfice tensionnel de la chirurgie dans les MACS unilatéral équivalente à celle des chirurgies d'adénome de Conn. Le bénéfice sur le contrôle glucidique semble possible dans les métaanalyses.
Le patient est opéré : faut-il le mettre sous hydrocortisone en post-opératoire ?
Réponse : Oui
Le % d'insuffisance corticotrope en post-opératoire augmente avec le degré d'hypercortisolisme mais est présent chez les patients MACS (jusqu'à 51 % dans une étude). Un bon prédicteur de l'insuffisance corticotrope est un cortisol après freinage supérieur à 5 ug/L (attention l'ACTH pré-opératoire n'est pas un bon prédicteur du risque d'insuffisance corticotrope).
À 2 ans post-opératoire, le patient avait une tension artérielle contrôlée sans anti-hypertenseurs, perdu 7 kg, une intolérance au glucose persistante mais n'a plus de dyslipidémie.

Léa CLOUAIRE
Docteur Junior EDN
Grenoble
D'après l'atelier de Julie SARFATI et Stéphanie ESPIARD