

Génétique de l'hyperaldostéronisme primaire : de la maladie rare à la maladie fréquente (Dr ZENNARO)
Pour commencer, on évoque une nouvelle définition plus nuancé de l'HAP comme une production d'aldostérone inappropriée, autonome par rapport au système rénine-angiotensine et non suppressible par charge sodée.
Selon les séries on retrouve jusqu'à 27 % d'HTA secondaire dans le service dont 11 % d'HAP sur une série de l'HEGP (M. Azizi, L. Amar), voire cela concernerait 5 à 6 % des patients hypertendus en soins primaires.
Il s'agit également d'une pathologie dont le diagnostic est complexe puisque inférieure à 2 % de patients à risque qui sont dépistés et inférieure à 1 % sont diagnostiqué permettant la mise en place d'un traitement approprié.
Depuis l'apparition de la génétique à haut débit, on a pu identifier des formes familiales notamment, la découverte d'HAP à des phases spécifiques de la vie comme pendant la puberté ou la grossesse.
Synthèse des formes familiales d'hyperaldostéronisme primaire :
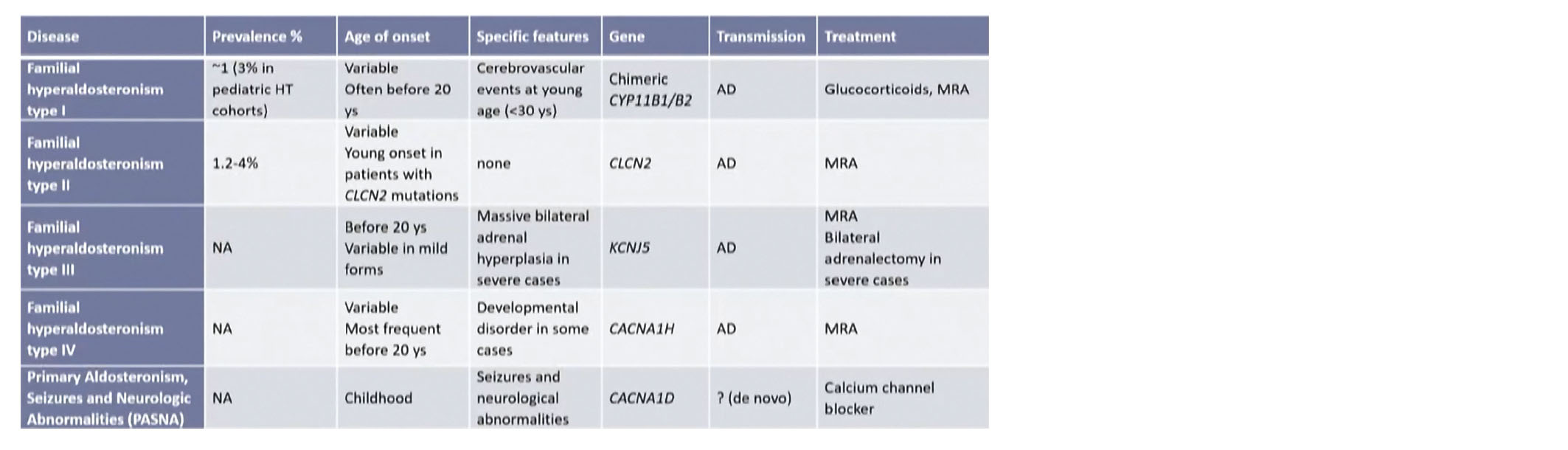
Ces formes familiales s'associent des particularités spécifiques qu'il convient de dépister. Seul le type 1 est suppressible par les glucocorticoïdes.
En dehors des formes familiales, on retrouve également des formes somatiques qui ne sont retrouvées que dans la partie adénomateuse de l'adénome de Conn (et pas dans la surrénale saine). Ces mutations touchent tous les gènes des formes familiales ainsi que d'autres gènes (codant pour les ATPases notamment), correspondant à supérieure à 90 % des adénomes de Conn.
Certaines de ses mutations somatiques ont également des particularités. Les mutations KCNJ5 représentent environ 40 % de nos patients en France, touchant des patients plus jeunes et la 1ère mutation chez la femme. La 2ème mutation la plus fréquente (jusqu'à 10 %) est CACNA1D, touchant préférentiellement des hommes d'âge plus avancé. Les mutations CTNNB1, plus fréquentes chez la femme, s'associent également à d'autres mutations (GNA11/GNAQ) avec développement à la grossesse, à la puberté ou à la ménopause, s'expliquant par une expression particulière de l'ARN (expression du récepteur au LHRH et hCG, le LHCGR).
Au niveau de la zone glomérulée, on retrouve une diminution de l'expression de cellules exprimant l'aldostérone synthase avec l'âge mais plutôt la présence de certains regroupement de cellules dans la glomérulée (APCC) sans adénome qui peuvent être la cible de mutations génétiques somatiques et vont être autonomes par rapport au reste de la surrénale.
Dans les données de recherche actuelle, on retrouve les gènes CASZ1 et RXFP2, qui, lorsqu'ils sont surexprimés en raison d'un polymorphisme, vont aboutir à la suppression de la production d'aldostérone sans affecter la production de cortisol. Parmi les mécanismes évoqués, on fait l'hypothèse qu'une diminution de la production d'aldostérone va stimuler de manière chronique la surrénale, via le SRAA, augmentant la production d'aldostérone, à terme cela stimulerait une prolifération cellulaire et donc un environnement propice à l'apparition de mutations somatiques.
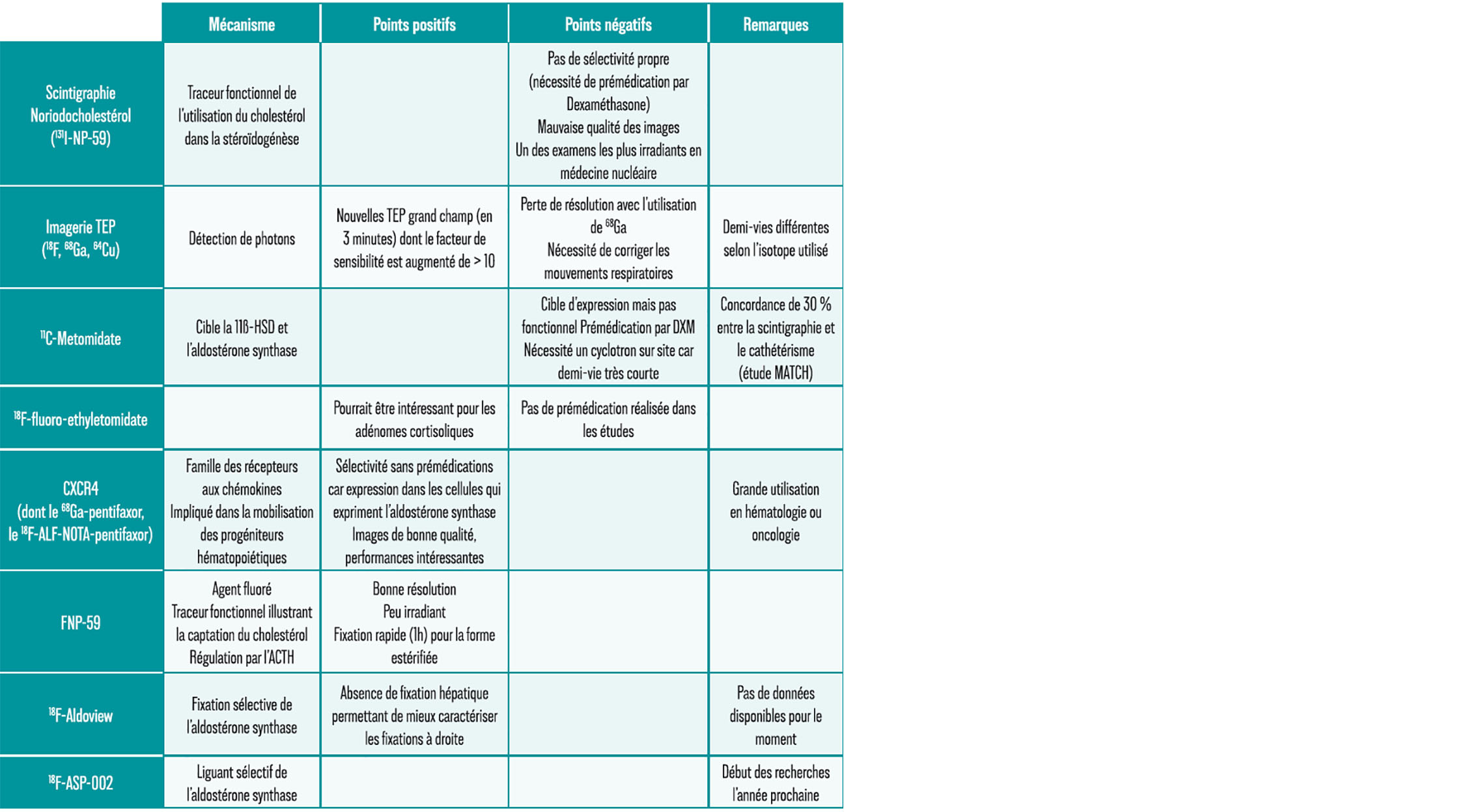
La place de la médecine nucléaire dans l'hyperaldostéronisme primaire (Pr TAIEB)
Il existe certaines limitations à l'imagerie puisque le foyer sécrétoire d'aldostérone peut être de petite taille. En ce qui concerne le cathétérisme veineux, cela reste une technique invasive, difficilement réalisable, protocoles différents et opérateur dépendant en faisant une exploration de centre très spécialisé.
La question de ce jour est donc : peut-on disposer d'un outil d'imagerie fiable, disponible, non invasif et qui pourrait caractériser les patients ?
Traitements médicamenteux de l'hyperaldostéronisme (Pr Michel BURNIER)
L'hyperaldostéronisme primaire (HAP) est fréquent en cas d'hypertension apparemment résistante au traitement (environ 20-30 % des cas) et s'associe à davantage de complications cardiovasculaires ou rénales que l'HTA essentielle.
Selon les guidelines de l'ESH le traitement semble simple : traitement chirurgical en cas d'adénome surrénalien unilatéral (adénome de Conn), traitement médicamenteux en cas d'atteinte surrénalienne bilatérale.
Dans la littérature, on retrouve beaucoup de données sur les traitements utilisés habituellement dans l'HAP, mais dans d'autres indications.
Notamment l'étude PATHWAY 2 qui évaluait les traitements dans l'HTA résistante et montrait que le médicament le plus efficace était la spironolactone.
Parmi les nouvelles thérapeutiques évoquées dans l'HAP on note les antagonistes non-stéroïdiens des récepteurs de l'aldostérone et les inhibiteurs de l'aldostérone synthase. Cependant ces thérapeutiques ont été investiguées essentiellement dans d'autres pathologies que l'HAP (diabète, insuffisance cardiaque, insuffisance rénale chronique).

Au total
Le traitement classique reste la chirurgie pour les atteintes unilatérales et les antagonistes stéroidiens des récepteurs aux minéralocorticoïdes pour les atteintes bilatérales.
De nouvelles approchent sont en cours de développement cependant peu de molécules sont étudiées spécifiquement dans l'hyperaldostéronisme primaire, notamment à grande échelle.

Mélissa BENALLOU
Interne EDN, 6ème semestre